NAMD Spotlights
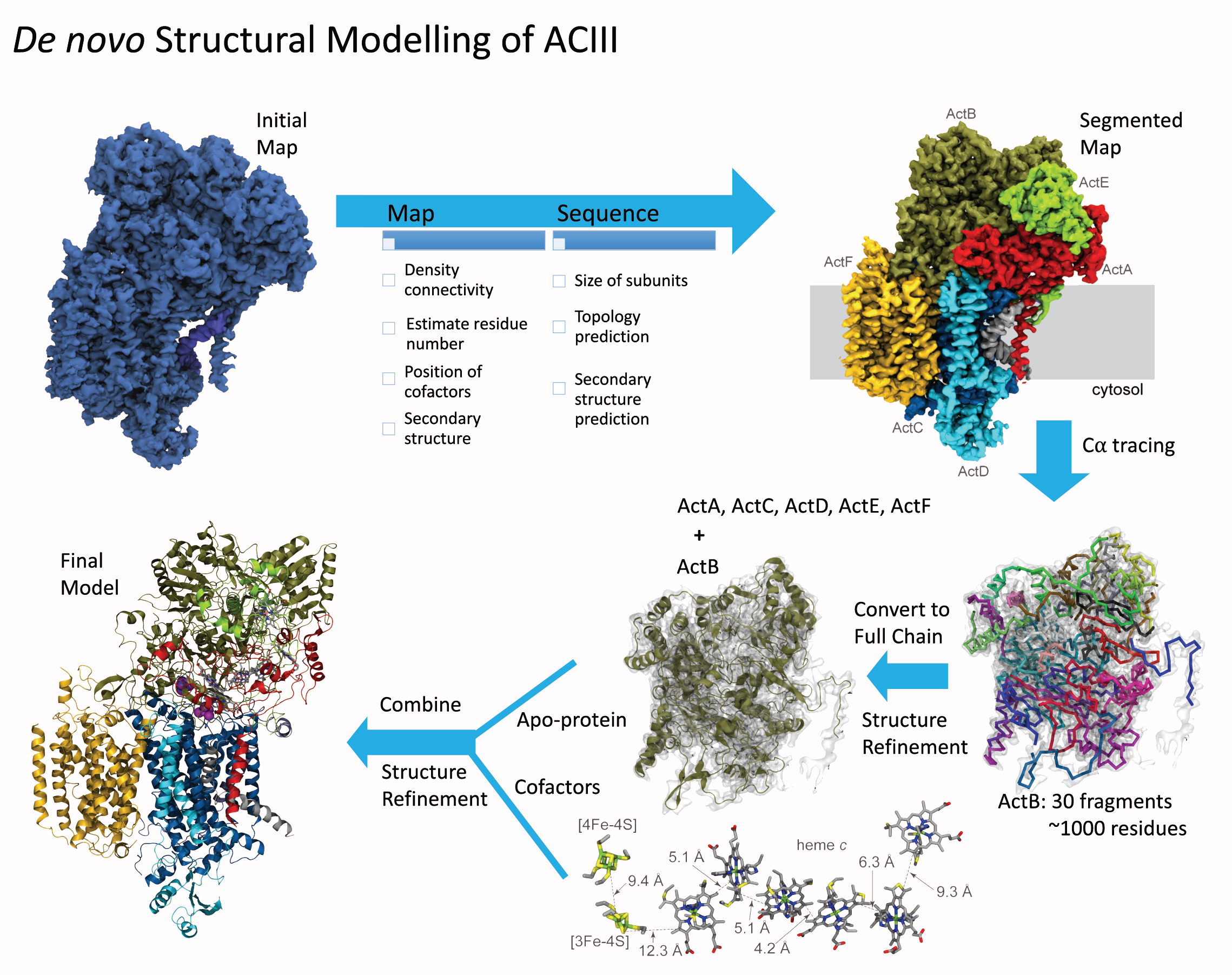
image size:
1.9MB
made with VMD
Bacteria are remarkable at tailoring their bioenergetic machineries to adapt to and thrive in diverse and ever-changing environments. A critical metabolic task is the efficient extraction of energy from food. Similar to us, many bacteria pass electrons to oxygen or other acceptors with the help of membrane-bound enzymes, and in doing so, they move protons across their membrane, thus generating a transmembrane voltage (much like a battery) that can be used for ATP synthesis. Typically, one enzyme passes an electron to its downstream enzyme via random collisions. However, sometimes these enzymes can form a supercomplex that positions the enzymes very close to each other and with the right pose for faster electron transfer and therefore more efficient energy conversion. In a recent paper reporting a three-way collaboration between biochemists, experimental structural biologists, and the Center researchers, the structure of one such supercomplex (termed Alternative complex III) was determined at an atomic resolution through a combination of cryoEM and advanced molecular modeling and simulation tools performed with NAMD and VMD. Read more about this study in Nature.

image size: 259.1KB
Staph infections are caused by bacteria commonly found on the skin of healthy individuals, where they can only cause relatively minor skin problems. However, when staph bacteria enter a person's bloodstream, they can travel to locations deep within the body causing infections that are often hard to treat. Not surprisingly, staph infections are the leading cause of healthcare-related, so called nosocomial infections. Particularly vulnerable areas are medical devices such as artificial joints or cardiac pacemakers, where the bacteria strongly stick to through formation of biofilms. Central to biofilm formation is an unusually tight interaction between microbial surface proteins called adhesins and the extracellular components of the host cells. In a recent report in Science , researchers used a combination of atomic force microscopy and GPU-accelerated molecular dynamics simulations using QwikMD and NAMD , to explore how the connection between adhesin and its target fibrinogen peptide can withstand huge forces greater than 2 nano-Newtons (see also Perspective in Science. ). The geometry and molecular details of the interaction ensures that, when pulled, the load is distributed over many hydrogen bonds all of which need to be broken at once before separating the bacterium from the surface of the host cell (see video on YouTube ). The unexpected, new mechanism, expands our understanding of why pathogen adhesion is so resilient and open new avenues for an intelligent design of antimicrobial therapies through development of anti-adhesion drugs. Read more in Science.
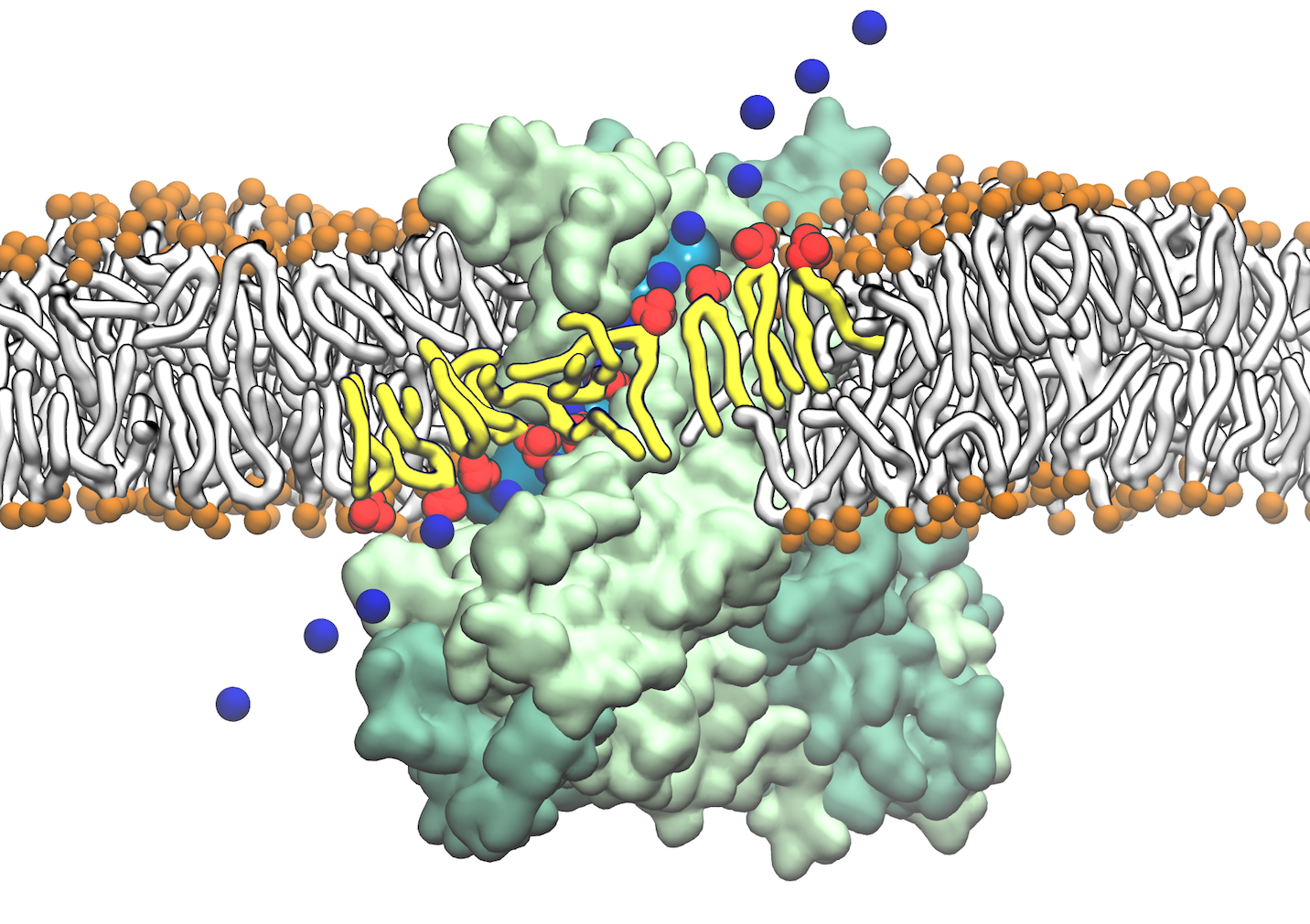
image size:
128.0KB
made with VMD
From bacteria to mammals, different phospholipid species are segregated between the inner and outer leaflets of the cellular membrane by ATP-dependent lipid transporters. Disruption of this asymmetry by ATP-independent phospholipid scrambling is a key step in cellular signaling, e.g., in apoptosis and in blood coagulation. Using molecular dynamics simulations with NAMD coupled with experimental assays, a recent collaborative study with H. Criss Hartzell (Emory University) published in eLife shows how a hydrophilic track formed on the surface of scramblases serves as the pathway for both lipid and ion translocations and how Ca2+ controls the open/closed transition of the track. The results of the simulations were used to successfully engineer scramblase activity in a homologous Ca2+ activated ion channel member of the family. This work provides a microscopic view of how lipids and other lipophilic molecules can permeate specialized proteins to travel between the two leaflets of the cellular membrane. Further details of the study can be found here.
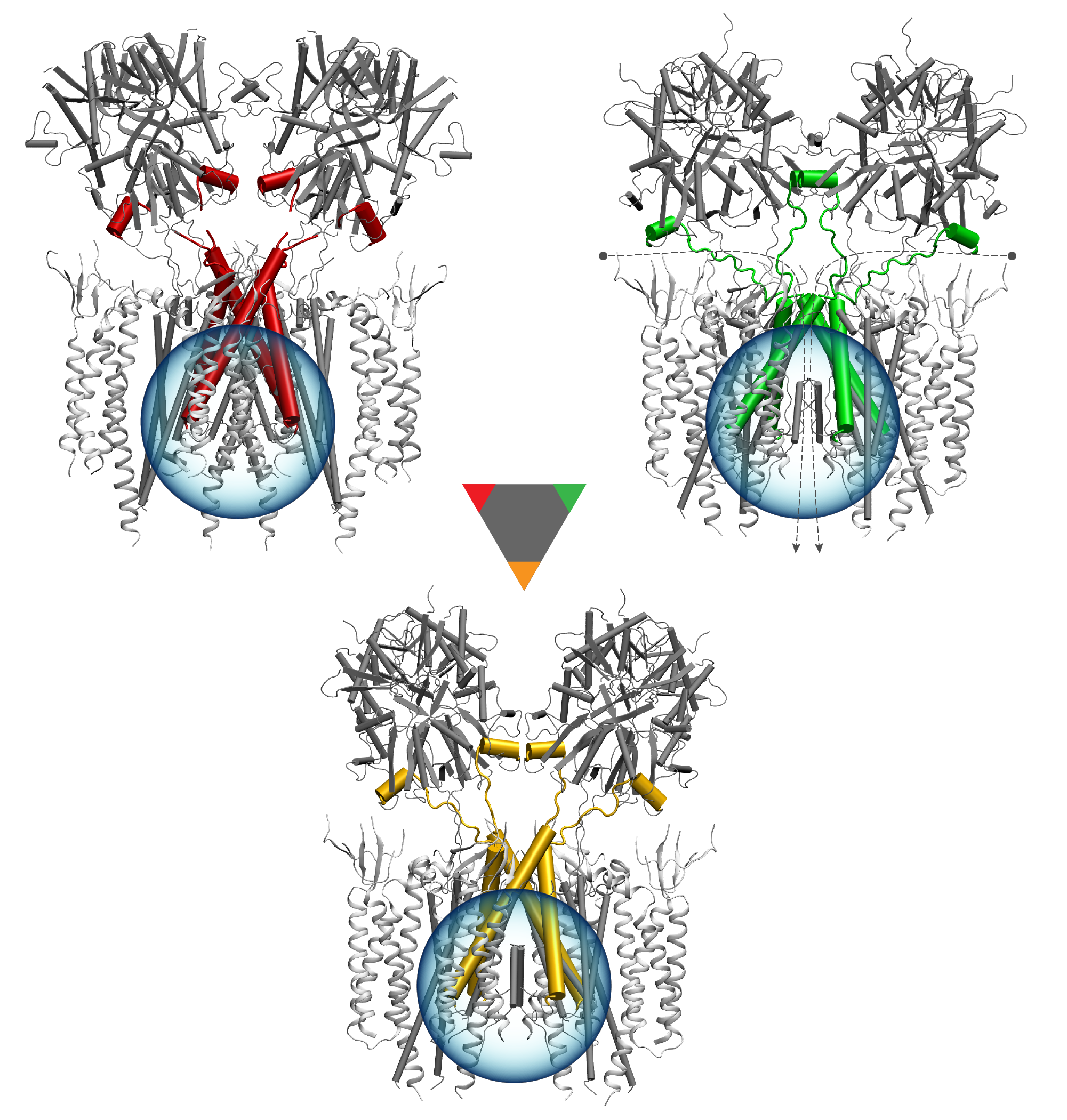
image size: 1.7MB
Deciphering the nuts and bolts of the how the brain works is crucial for understanding human psychology and behaviors developing novel and more effective treatments for neurological diseases. The human brain's function relies on the transmission of electric signals from one neuron to another through the synapse, a delicate process mediated by diffusion of neurotransmitter molecules released from the presynaptic cell that bind their specific receptor on the postsynaptic cell. In a recent collaborative study, performed with the experimental lab of Eric Gouaux (Vollum Institute), one of the key receptors, known as the AMPA receptor, was characterized in its three major functional states: closed, desensitized and active (see Figure 1). The study has uncovered the long-sought structures for the active and desensitized states of this protein for the first time (using cryo-EM) after decades of research. These structures provide vital information as to how this ion channel receptor converts the chemical signal of the neurotransmitter to a temporally controlled excitation profile in the postsynaptic neuron. The method of MDFF developed by the Center was used for refinement of the structure, and NAMD simulations together with VMD visualization were used to characterize the open state of the channel.
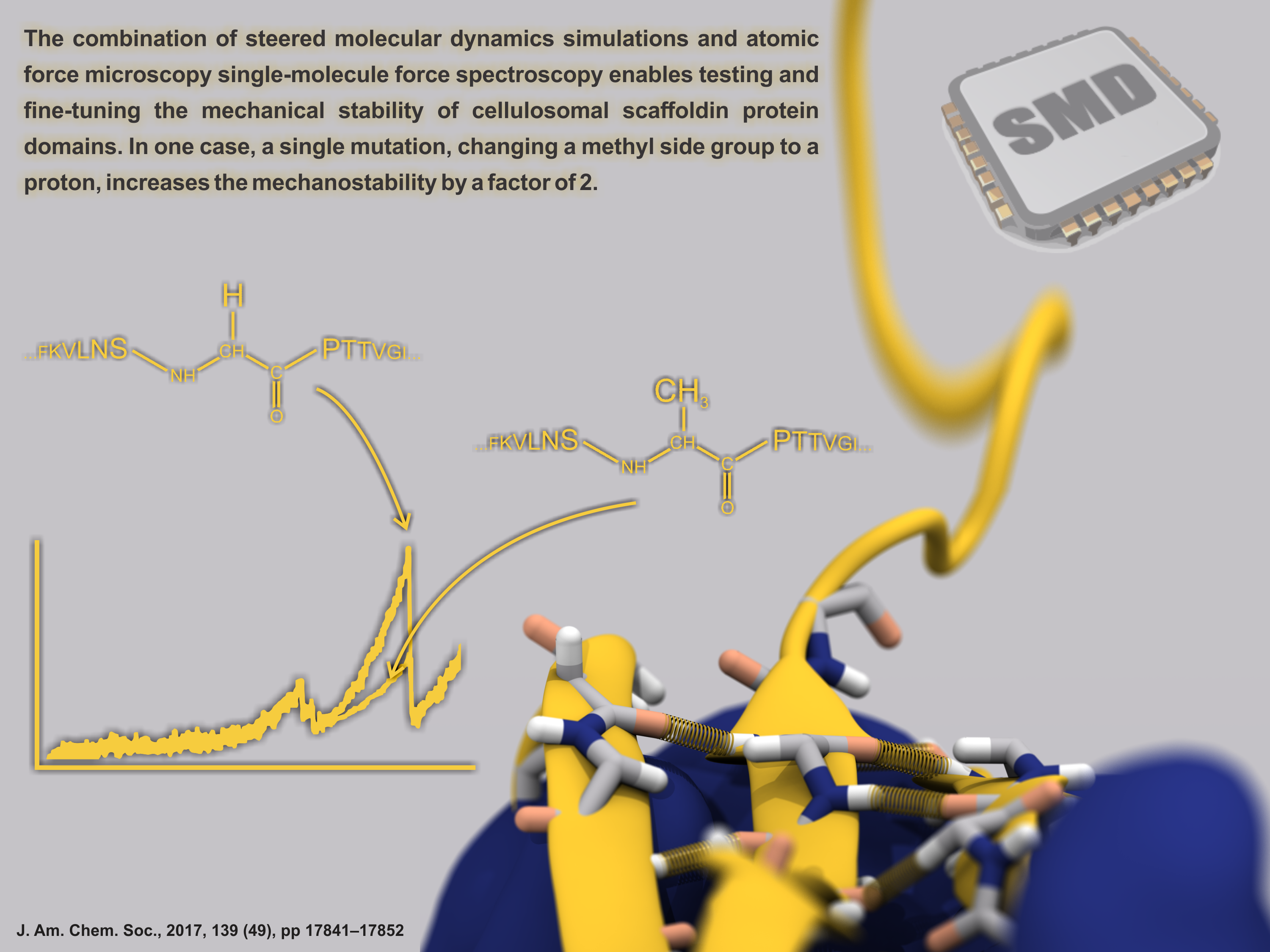
image size: 303.2KB
Proteins play major mechanical roles in a cell. Mechanical properties of proteins can be substantially modulated by slight changes in their amino acid composition, an ability essential to bacterial cells, which use protein repeats with small modifications to tune their mechanical strength, and thus their functional properties. A prime example of the role of biomechanics in the cell is found in symbiont gut bacteria who need to hold to their food source in such turbulent environments as the rumen of the cow. Proteins evolved for this purpose are known actually to be among the strongest mechanical biomolecules. Key to the process are molecular tentacles, so-called cellulosomes, on the surface of symbiotic bacteria. The cellulosomes develop a tight grasp on and then effective cleavage of hardy cellulose fibers of the grass. In a joint experimental-computational study researchers investigated the mechanical properties of cohesin, a major cellulosome component, under force. Using NAMD, all-atom steered molecular dynamics (SMD) simulations on homology models offered insight into the process of cohesin unfolding under force. Based on the differences among the individual force propagation pathways and their associated correlation communities, the researchers were capable of designing protein mutants to tune the mechanical stability of the weakest cohesin. The proposed mutants were tested with high-throughput atomic force microscopy experiment revealing that in one case a single alanine to glycine point mutation suffices to more than double the mechanical stability. Read more about our cellulosome research here.
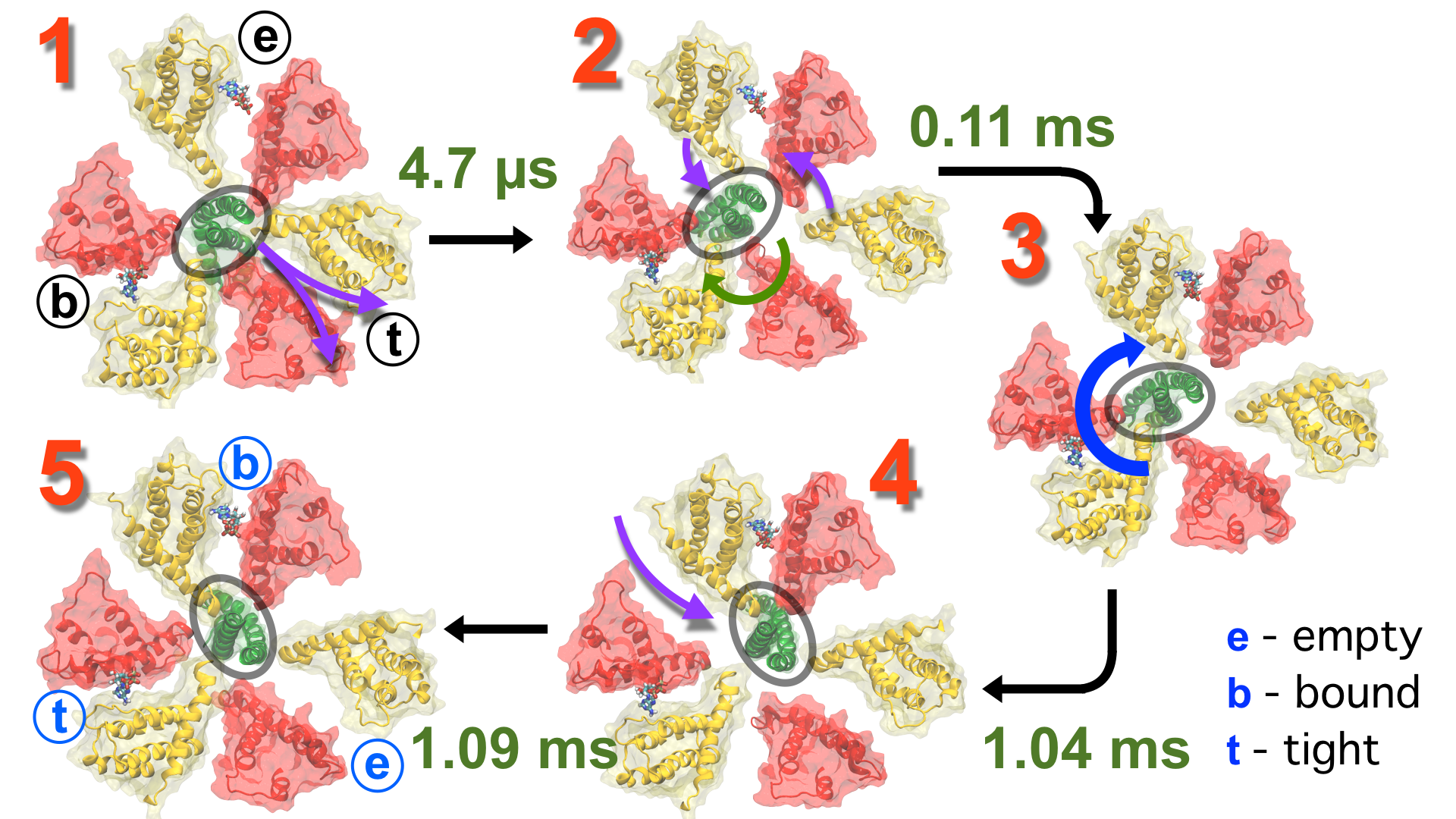
image size:
1.7MB
made with VMD
Living cells employ an intricate network of biochemical processes for the conversion of solar energy or nutrients into energy-rich Adenosine Tri Phosphate (ATP) molecules. From bacteria to fungi, plants, and animals, ATP serves as the universal energy currency of life, fueling the processes cells need to survive and function. Over the course of a day, an individual will typically use the equivalent of his or her bodyweight in ATP; however, the human body carries only a small amount of the molecule at any one time. That means cells must constantly recycle or replenish the ATP molecules, relying on a highly efficient motor protein called ATP synthase to do the job. Given its ubiquitous role in photosynthesis and respiration, efficiency of the ATP synthase motor has been a focus of intense biochemical investigations over the past three decades, which included Boyer and Walker's Nobel Prize-winning contributions in 1997. Yet, connection between the Chemistry of ATP dissociation and Physics of ATP synthase motor-action remained elusive. A recent study based on molecular dynamics simulations with NAMD reveals the working principles of V-type ATP synthase in atomic resolution. Swiveling motions in the protein ring were captured together with rubber band-like elasticity of the motor's central stalk. This swiveling motion of the ring when paired with the stalk absorbs about 75 percent of the energy released during ATP hydrolysis, showcasing a molecular design that underlies the molecular motor's remarkable energy-conversion efficiency. More on ATPase here and here.
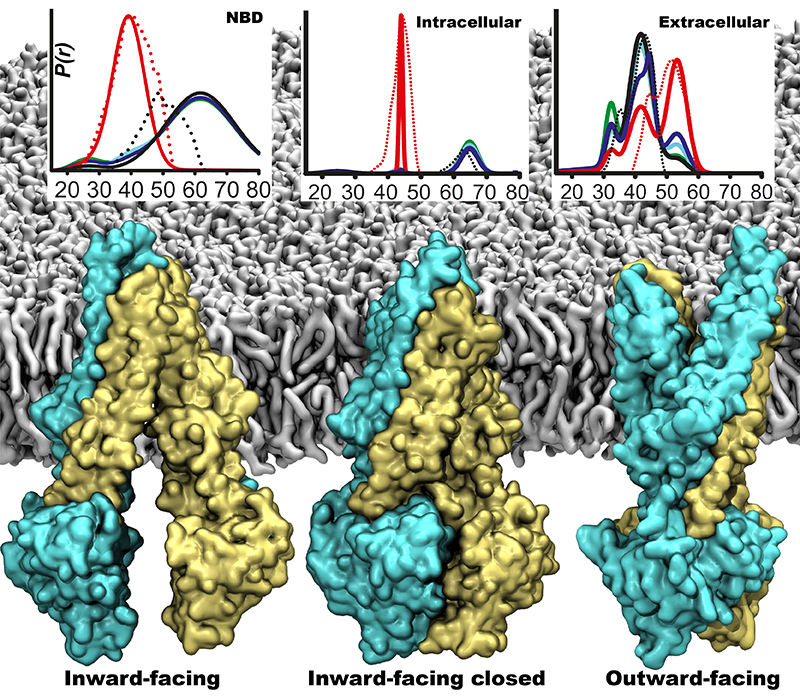
image size:
358.3KB
made with VMD
One of the most common mechanisms by which cancer and microbial cells develop resistance against chemotherapeutic agents is to express a large number of specialized transporter proteins in their cellular membrane that use the universal cellular energy of ATP to actively pump the drug molecules to the outside. P-glycoprotein, a prominent member of such molecular "vacuum cleaners" and responsible for multidrug resistance (MDR) in a wide variety of cancer types, accomplishes its role by undergoing large-scale structural transitions in the cellular membrane through which it effectively moves drug molecules from one side of the membrane to the other. In a recent collaborative publication in Nature with leading experimental groups at Vanderbilt and Virginia, and employing advanced molecular modeling and simulation techniques implemented in NAMD, a robust structural model was developed for the unknown outward-facing state of P-glycoprotein, allowing a full structural description of the transport cycle, and a novel mode of energy transduction. Further details of the study can be found here.
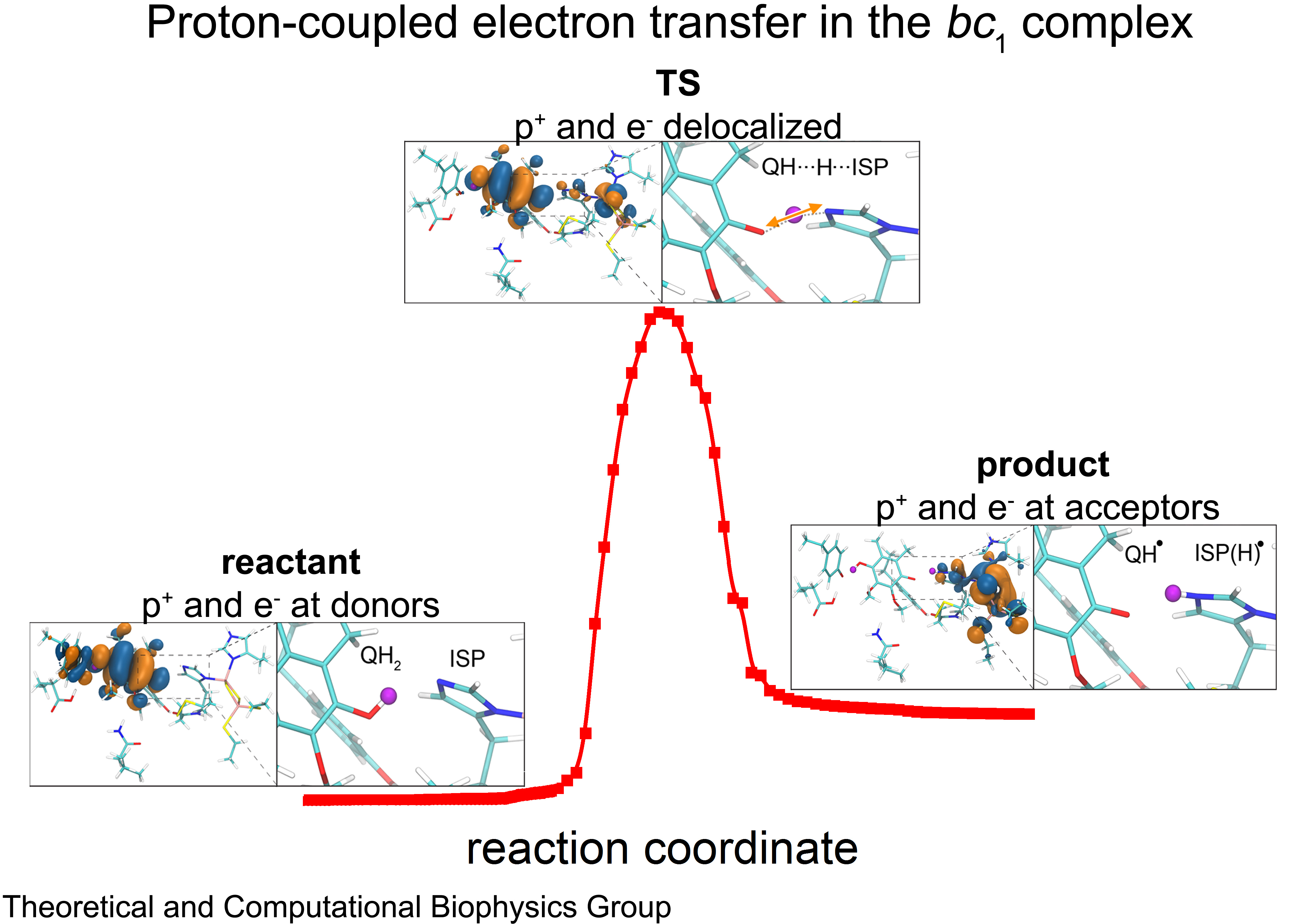
image size:
1.5MB
made with VMD
Cellular respiration and photosynthesis are the primary energy production mechanisms for sustaining life on earth. Both processes use input energy (food or sunlight) to drive coupled electron and proton transfer reactions, thus replenishing the electrical charge of the cellular membrane, which in turn is used to produce ATP - the universal fuel for all cellular activities. A key step involved in both bacterial photosynthesis and mitochondrial respiration is mediated by a specialized protein complex, the bc1 complex, which seizes the energy released from interconversion of quinol and quinone to pump protons across the bioenergetic membrane. A recent study, combining molecular simulations performed with NAMD and quantum chemical calculations, unveiled the coupled nature of the proton and electron transfer reactions in the quinol binding site of the bc1 complex from a photosynthetic bacterium at an unprecedented level and described the role of key mechanistic elements. More about the bc1 complex can be found here.
image size:
507.7KB
made with VMD
The 2.12 release of the molecular dynamics program NAMD provides major enhancements in performance, flexibility, and accuracy, complementing the greatly enhanced usability provided by the QwikMD GUI released in VMD 1.9.3. NVIDIA GPU-accelerated simulations with NAMD 2.12 are up to three times as fast as 2.11, particularly for implicit solvent simulations and single-node simulations of smaller systems. NAMD 2.12 is also optimized for the new Intel Xeon Phi KNL processors found in Argonne Theta, NERSC Cori, and TACC Stampede 2. NAMD 2.12 builds on the asynchronous multi-copy scripting capabilities introduced in NAMD 2.11 with the ability to modify and reload the molecular structure, enabling development of grand canonical and constant pH ensemble methods, as well as an optional Python interface for advanced on-the-fly analysis. Finally, NAMD 2.12 provides a complete, no-recompilation-needed interface for hybrid QM/MM with both the semi-empirical code MOPAC and the ab initio/DFT code ORCA. More on new features in the 2.12 release of NAMD can be found here. NAMD is available free-of-charge as source code, precompiled binaries, pre-installed at supercomputer centers, and now jointly with VMD as one-click interactive molecular modeling on the Amazon cloud.

image size:
2.2MB
made with VMD
The latest release of VMD brings many advances that help researchers prepare, analyze, and visualize molecular simulations. The new QwikMD plugin streamlines key simulation preparation and analysis tasks, and guides users in the creation of reusable simulation workflows and protocols. VMD now includes several advanced features for parallel analysis and visualization of cellular-scale simulations, as reported here, and here. VMD 1.9.3 strengthens collaboration between experimental and computational biologists by supporting a broader range of experimental density map image formats, such as those used in cryo-electron tomography. Many updated plugins are included in VMD 1.9.3, including tools for analysis of free energy perturbation simulations, MDFF hybrid structure fitting, ffTK force field parameterization, and normal mode analysis. VMD 1.9.3 adds support for new hardware and operating system platforms including IBM OpenPOWER (ORNL Summit), a variety of GPU-accelerated ARM SoCs, the Amazon AWS EC2 cloud, and most recently, the Intel Xeon Phi Knight's Landing many-core CPU (TACC Stampede 2, Argonne Theta). The VMD 1.9.3 release adds stunning graphics produced using interactive ray tracing using the latest multi-core CPUs and GPU accelerators, enabling 360-degree panoramic movie rendering for VR headsets, as reported here, and here. Interactive ray tracing makes the task of getting a molecular image "just right" much easier than ever before; it also enables rendering of spectacular movies for communication of scientific results. A VR movie rendering tutorial assists users with the steps required in rendering and encoding VR movies for upload to YouTube for display using VR headsets such as Google Cardboard, Oculus Rift, and GearVR. More details about VMD 1.9.3 features can be found here.
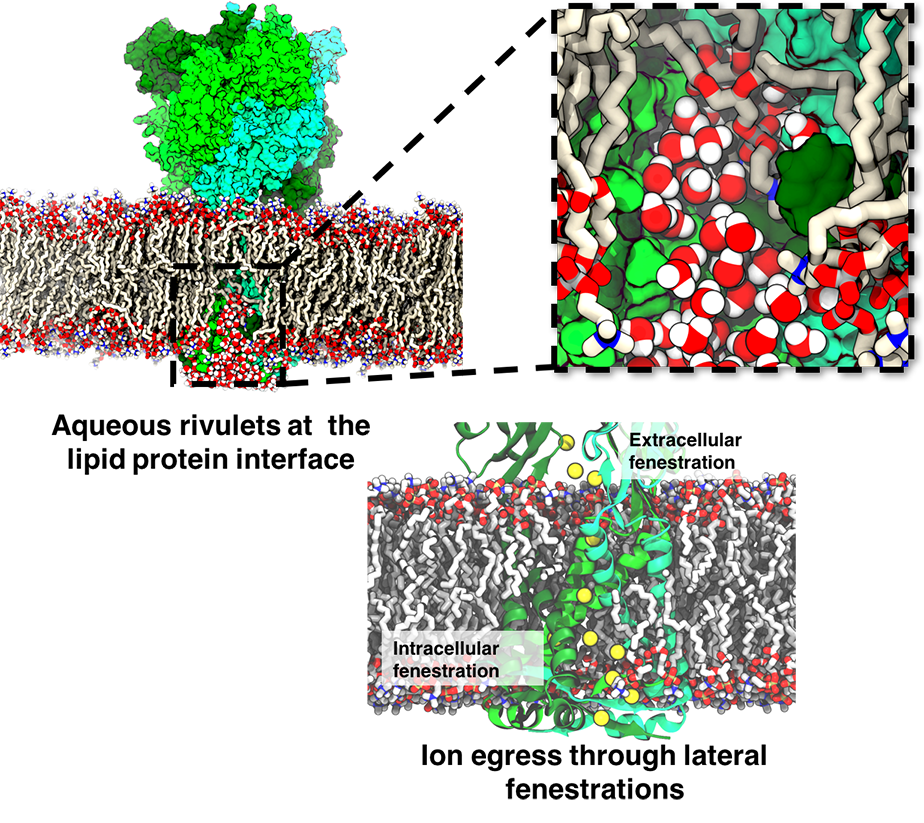
image size:
792.2KB
made with VMD
ATP, a ubiquitously prevalent biomolecule, which is best known for being the principal energy source for a living cell, also plays a crucial role in inter-cellular communication, thus acting as a signaling molecule. One of the major receptors in this signaling cascade are the P2X receptors which are trimeric, non-selective cation channel activated by ATP and responsible for key processes such as muscle contraction, inflammatory response, pain, and even taste signal transduction. As a result of their extensive prevalence and important implications in human physiology, P2X receptors serve as important pharmacological targets for cardiovascular, neuronal, and inflammatory diseases. In a recent collaborative study with experimental structural biologists, molecular dynamics simulations of a membrane-embedded model of a P2X receptor performed with NAMD were used to reveal intricate details of the ion permeation mechanism and pathway. Surprisingly, it was observed that one half of the ion permeation pathway is composed of lipids on one side and of the protein residues on the other side, a novel design for an ion translocation pore. The study demonstrates yet another active functional role for lipids in membrane protein function, further emphasizing the importance of lipid protein interactions in biological processes. More details can be found here.
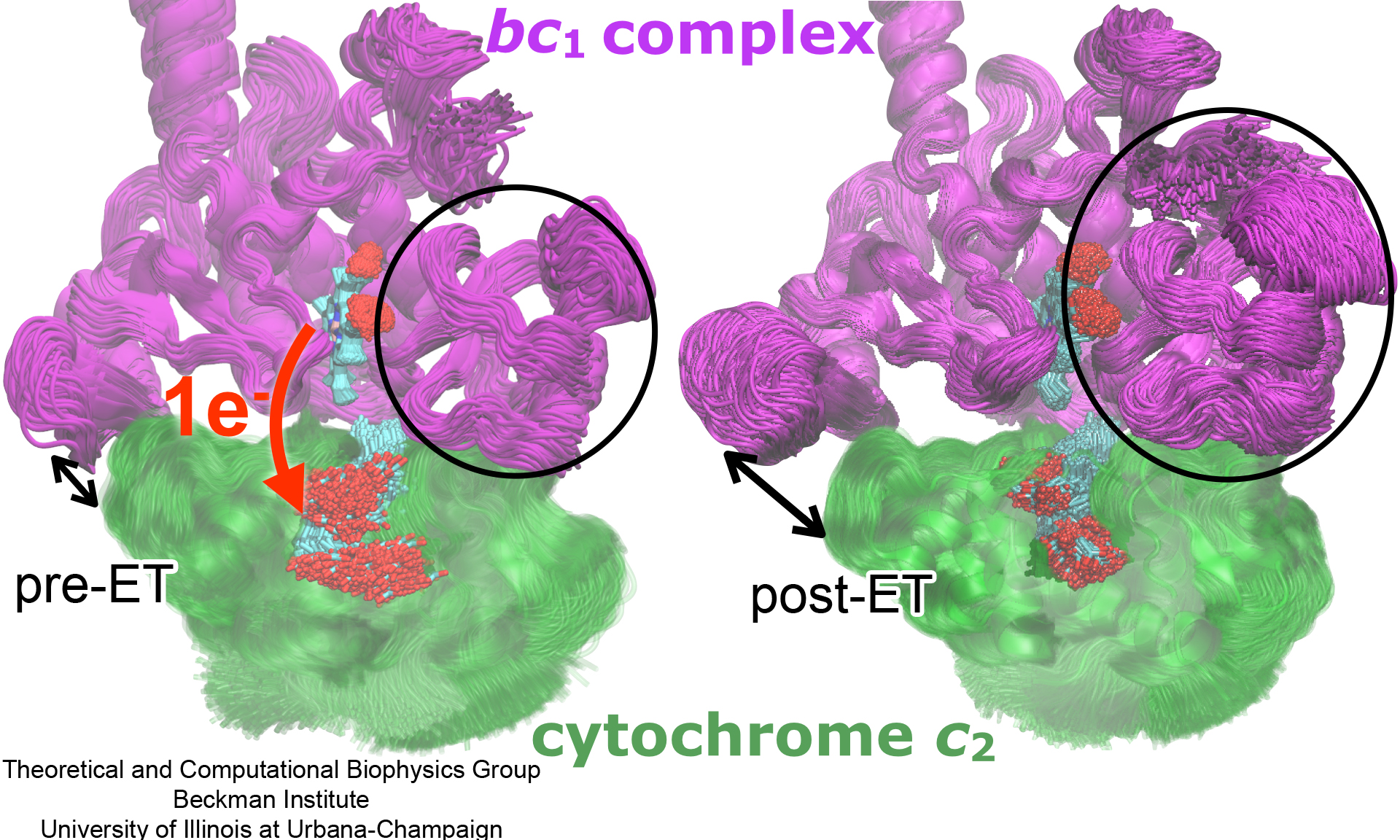
image size:
1.6MB
made with VMD
Photosynthetic organisms have been optimized by over two billion years of evolution into energy-harvesting machines that surpass the efficiency of man-made solar devices. Employing a network of hundreds of proteins, these organisms transform energy from the incident sunlight into ATP molecules - the universal fuel for sustaining cellular activities. A key step in the conversion of solar energy into ATP involves shuttling of negative charges or electrons between widely separated sites on the cell membrane. This membrane-wide charge transportation is accomplished by the cytochrome c family of proteins. Cytochrome c finds and docks to an electron donor protein, accepts the electron and unbinds as a result of it, and afterwards finds an electron acceptor protein, docks to it to deliver the electron and unbinds to repeat the sequence. Given its ubiquitous role in photosynthesis and respiration, the recognition, binding and unbinding mechanism of cytochrome c have been a focus of intense biochemical investigations over the past three decades. However, little is deciphered on the details of cytochrome c activity. A recent study based on molecular dynamics simulations with NAMD reveals the working principles of cytochrome c in atomic resolution. The calculations suggest that electrostatic forces drive the cytochrome c binding, and enable membrane-wide electron transfer. More about the cytochrome c protein can be found here.
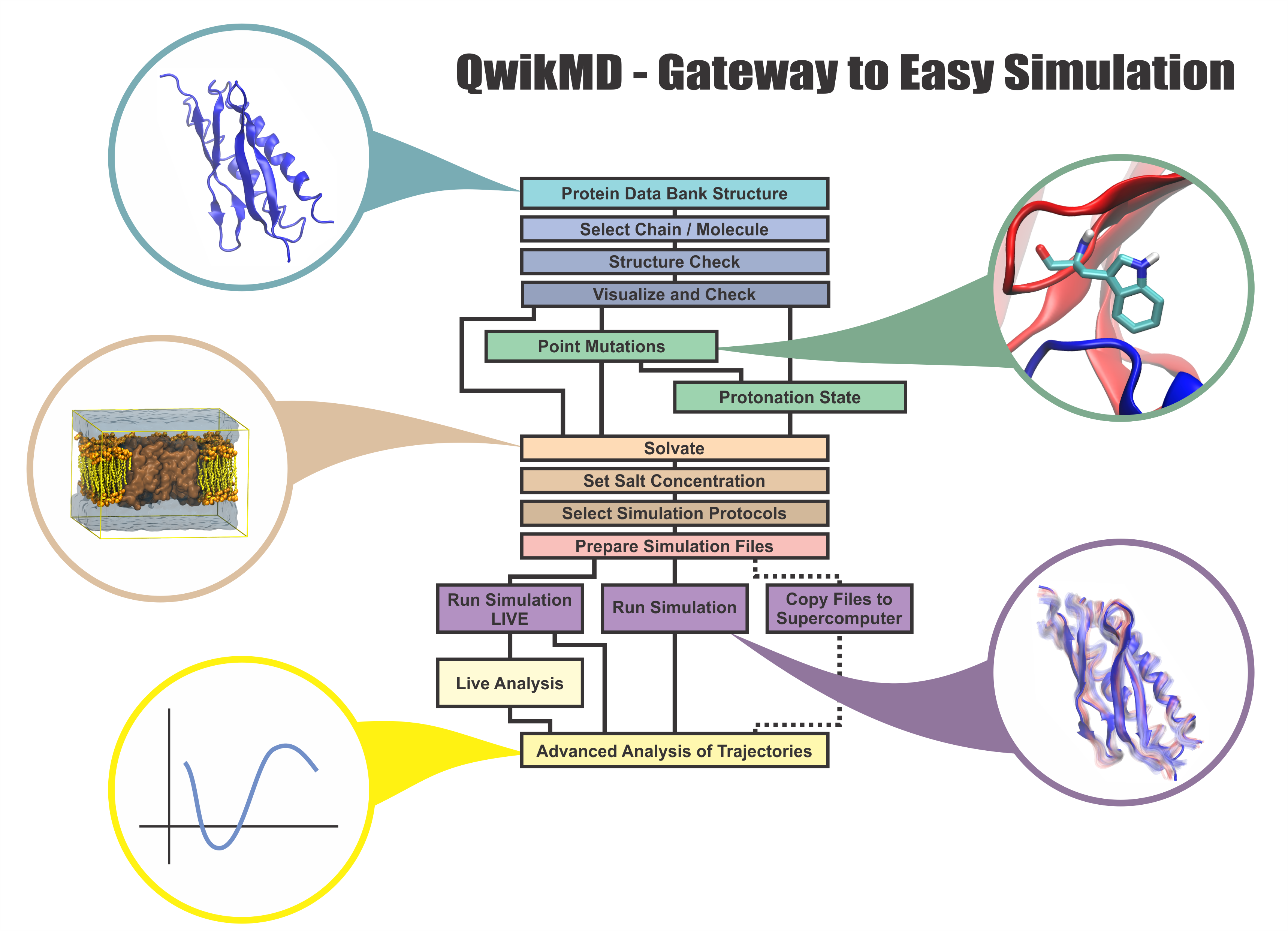
image size:
1.2MB
made with VMD
Everything that living things do can be understood in terms of jigglings and wigglings of atoms.
Richard Feynman's remark in the early 1960's summarizes what is today widely accepted, namely,
that molecular processes can be described by the dynamics of biological molecules, therefore connecting
protein dynamics to biological function. Molecular dynamics (MD) is by far the best tool to investigate
jigglings
and wigglings
of biological systems. Advances in both software and hardware
have spread the use of MD, however the steepness of the learning curve of the methodology of MD
remains high. To assist new users in overcoming the initial barrier to use MD software, and to help the more advanced users to
speed up tedious steps, we have developed the QwikMD software, as decribed in a recent paper.
By incorporating an easy-to-use point-and-click user interface that connects the widely used molecular graphics
program VMD
with the powerful MD program NAMD,
QwikMD allows its users to prepare both basic and advanced MD simulations in just a few minutes. At the same time,
QwikMD keeps track of every step performed during the preparation of the simulation, allowing easy reproducibility
and shareability of protocols. More information about QwikMD, as well as introductory tutorials are available on our
QwikMD webpage.
QwikMD is available in VMD 1.9.3 or later versions.
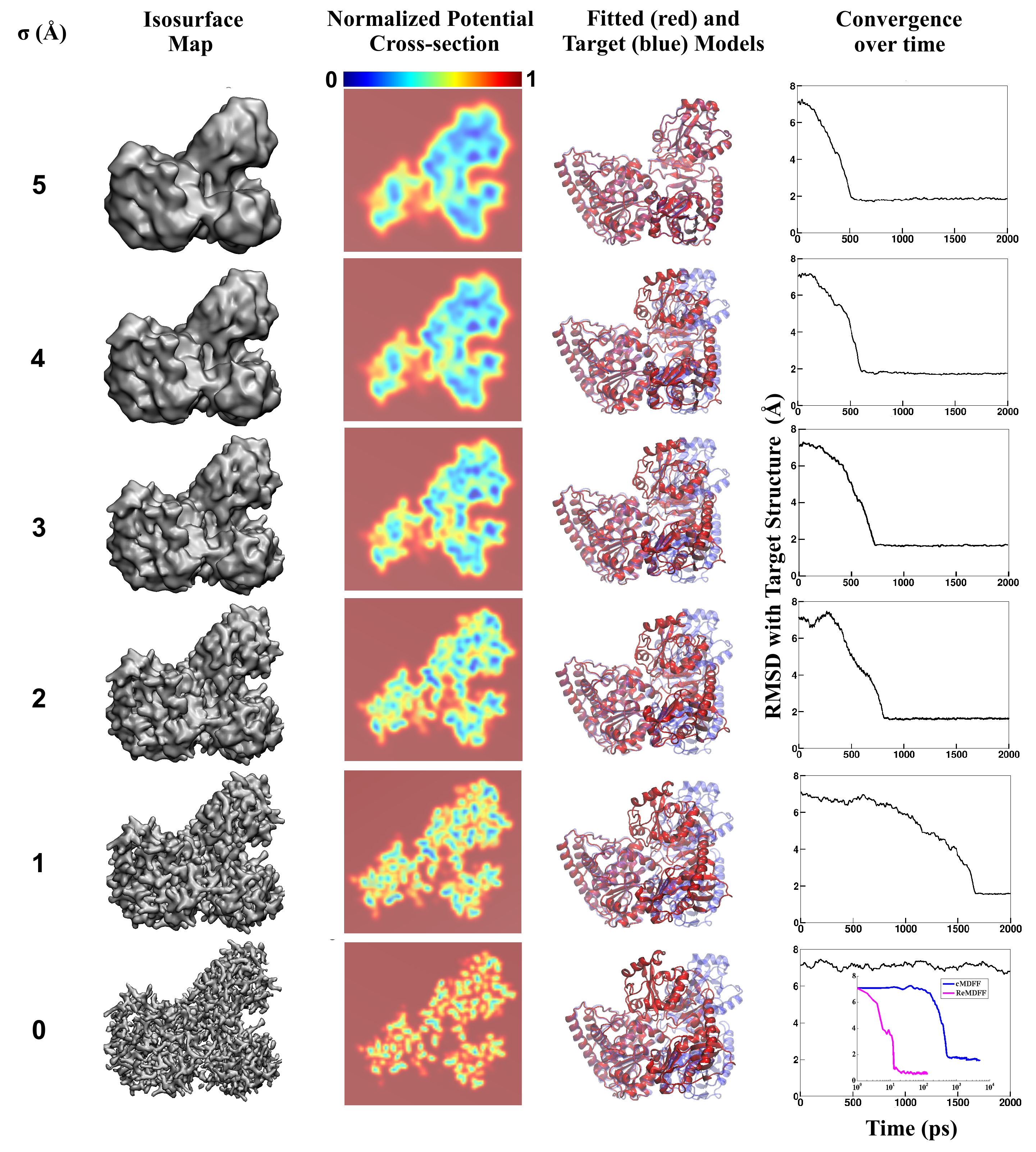
image size:
3.3MB
made with VMD
Living cells are brimming with the activity of macromolecular complexes carrying out their assigned tasks. Structures of these complexes can be resolved with cryo-electron microscopy (cryo-EM), wherein the complexes are first freeze-shocked into states characterizing their action and subsequently imaged by detection cameras. Recent advances in direct detection camera technology enable today's cryo-EM laboratories to image the macromolecular complexes at high-resolution, giving us a better view of the cell than ever before. Computational techniques like molecular dynamics flexible fitting (MDFF) are a key tool for producing atomic models of the imaged molecules, providing greater insight into their structure and function. The increased resolution of EM maps, which contain sharp valleys capable of trapping structures, presents a challenge to MDFF which was originally developed for maps in a lower resolution range. However, a recent study unveils two new techniques called cascade (cMDFF) and resolution exchange (ReMDFF) molecular dynamics flexible fitting to overcome the hurdles posed by high-resolution maps. The refinement is achieved by interpreting a range of cryo-EM images, starting with an image of fuzzy resolution and progressively improving the image's contrast until near-atomic resolution is reached. These techniques were employed to solve the structure of the proteasome, the recycling machine of the human cell. New analysis schemes that look at the flexibility of the obtained structure provide a measure of model uncertainty within the near-atomic EM images, improving their contrast. All the tools are available on cloud computing platforms allowing community-wide usage at low monetary cost; the complex computations can now be performed at the cost of a cup of coffee.

image size:
299.6KB
While waste recycling in daily life has become popular only recently, living cells have been recycling their protein content since the very beginning. Recycling of unneeded protein molecules in cells is performed by a molecular machine called the proteasome, which cuts these proteins into smaller pieces for reuse as building blocks for new proteins. Proteins that need to be recycled are labeled by tags made of poly-ubiquitin protein chains. The proteasome machine recognizes and binds to these tags, pulls the tagged protein close, then unwinds it, and finally cuts it into pieces. Despite its substantial role in the cell's life cycle, the proteasome's atomic structure and function still remain elusive. In our recent study, we obtained an atomic structure of the human 26S proteasome by combining computational modeling techniques, through molecular dynamics flexible fitting (MDFF) of the cryo-electron microscopy (cryo-EM) data. The features observed in the resulting structure are important for coordinating the proteasomal subunits during protein recycling. One of the key advances is that for the first time the nucleotides bound to the ATPase motor of the proteasome are resolved. The atomic resolution of the structure permits to perform molecular dynamics simulations to investigate the detailed proteasomal function, in particular the protein unwinding process of the ATPase motor. Furthermore, our obtained structure will serve as a starting point for structure-guided drug discovery, developing the proteasome as a crucial drug target. The atomic models are deposited in the protein data bank (PDB) with the PDB IDs 5L4G and 5L4K and the 3.9 Å resolution cryo-EM density is deposited in the electron microscopy data bank EMD-4002. More information about our proteasome projects is available on our proteasome website. Easy access to our modeling techniques is provided through QwikMD, which was employed here for the first time.

image size:
1.7MB
made with VMD
Our lungs are coated with a layer of protein and lipid mixture called lung surfactant, which prevents the lungs from collapsing and protects us from bacterial and viral infections (see October 2012 and January 2014 highlights). Lung surfactant protein A (SP-A) - the major protein constituent of lung surfactant - plays a dual role. It aggregates DPPC lipid, a major component of lung membrane, into a lattice-like structure that prevents the lungs from collapsing. SP-A is also known to recognize and bind bacterial lipids, namely lipid A, on surfaces of gram-negative bacteria, thereby helping to initiate various clearance mechanisms. However, it was unclear how SP-A exhibits such functional duality with its binding to two different types of lipids. A recent study used molecular dynamics simulations with NAMD to unravel the dual role of SP-A. Combined with crystallographic and mutational analyses, researchers have discovered several critical, non-canonical lipid binding sites that involves cation-π interactions and hydrogen bonds. Simulations have also revealed that SP-A binds stronger to bacterial lipid (lipid A) than to surfactant lipid (DPPC lipid), which suggests SP-A may prioritize its host defense functions by transferring from lung membrane to bacterial surface. These findings in atomistic detail will enable experimentalists to enhance the antimicrobial function of SP-A. More on our lung surfactant protein website.
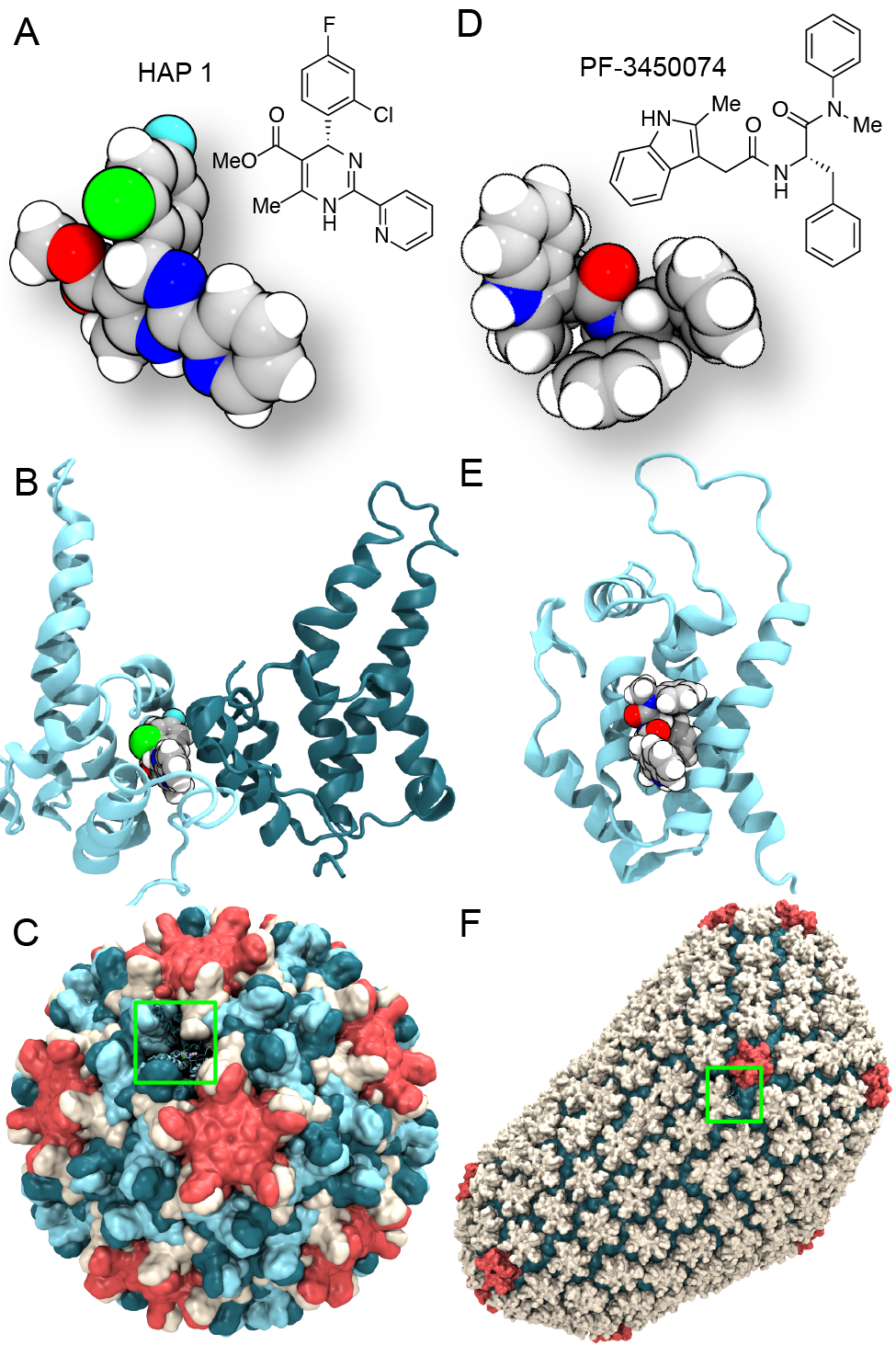
image size:
0 bytes
made with VMD
Virus capsids, specialized protein shells that encase the genome of viral pathogens, play critical roles in regulating viral infection, and are, thus, of great pharmacological interest as drug targets. In particular, small-molecule drugs (typically <900 Da) represent a promising antiviral strategy, and a number of such compounds have been developed to inhibit virus capsids by interfering with their assembly and uncoating processes. Importantly, to explain the mechanisms by which drugs disrupt capsid function, as well as to successfully design novel therapeutics, the interactions of drug molecules with their capsid targets must be studied at full chemical detail. Toward this end, all-atom molecular dynamics simulations are emerging as an essential technique to investigate the effects of small-molecule drugs on capsid structure and dynamics. Research presented in a recent Perspective applying simulations in NAMD to study drug-bound hepatitis B virus (HBV) and human immunodeficiency virus type 1 (HIV-1) capsids suggests the types of valuable chemical and physical information computational approaches can reveal, and underscores the importance of simulating, not isolated capsid proteins, but functional assemblies up to the level of complete capsids. Notably, through analysis with VMD, the study found that binding of the drug HAP1 to the HBV capsid causes global structural changes that subtly alter the overall capsid shape, including a flattening of capsid curvature. Further, the study found that the binding of the drug PF74 to the HIV-1 capsid imposes rigidity and causes shifts in allosteric communication pathways connecting distant regions of the capsid protein. The authors of the Perspective anticipate that many other such exciting discoveries regarding virus capsid function and their use as drug targets lie just ahead on the horizon, and molecular dynamics simulations will drive these discoveries pending a series of notable advancements in computational methodology.
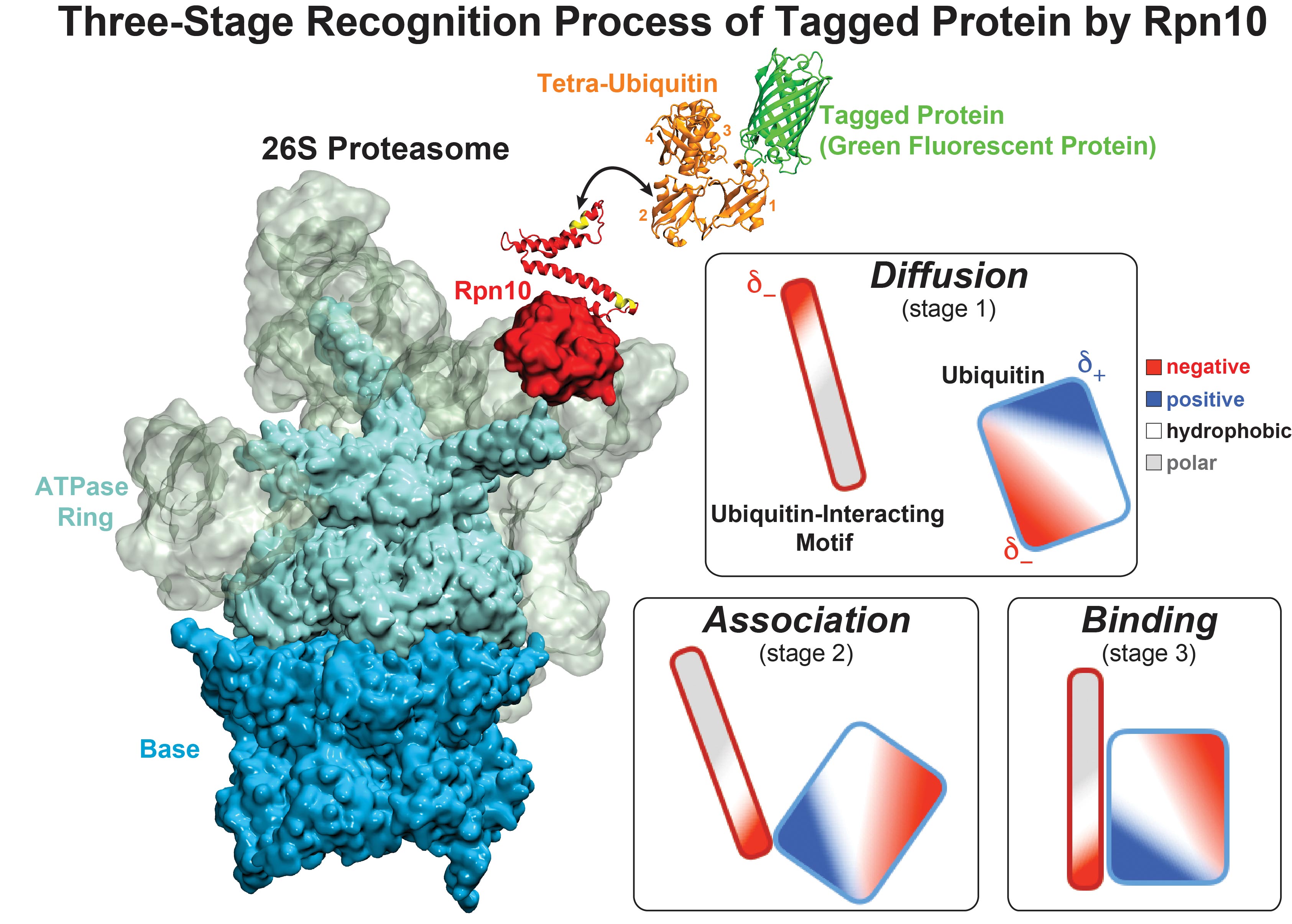
image size:
711.1KB
made with VMD
While waste recycling became popular in our daily life, living cells mastered waste recycling of their protein content since their very beginning. Recycling of unneeded protein molecules in cells is performed by a molecular machine called 26S proteasome, which cuts these proteins into smaller pieces and releases the pieces into the cell interior for reuse as building blocks for new protein. Proteins that need to be recycled are usually those that are misfolded. Proteins are recognized as such by the cells' so-called quality control system. This system labels misfolded proteins by a tag made of tetra-ubiquitin protein chains. The 26S proteasome machine recognizes and binds to these tags via its subunit Rpn10. After Rpn10 binds to the tetra-ubiquitin tag and pulls the protein close, the 26S proteasome unwinds the tagged protein and cuts it into pieces. A recent study, based on molecular dynamics simulations with NAMD, sheds light onto how 26S proteasome and Rpn10 recognize the tetra-ubiquitin tag in three stages: In stage 1 of the recognition process conserved complementary electrostatic patterns of Rpn10 and ubiquitins guide protein association; stage 2 induces refolding of Rpn10 and tetra-ubiquitin tag; stage 3 facilitates formation of hydrophobic contacts between the tag and Rpn10. More information is available on our 26S proteasome website.

image size:
386.7KB
made with VMD
RNA molecules are continuously synthesized in living cells as carriers of biological information written in the sequence of basic RNA units, called nucleotides. To keep cells healthy, RNA molecules not longer needed or with errors have to be removed. A large barrel-like protein complex, the RNA exosome, is a molecular machine that degrades unneeded RNA molecules, pulling them inside its long internal channel and cutting them sequentially into single nucleotides. A new molecular dynamics study, employing NAMD, shows that a special active protein subunit of the exosome, called Rrp44, grips tightly the RNA molecule throughout its extended channel. Rrp44 grips RNA molecules with five or more nucleotides in length while their ending nucleotides get sequentially cut, whereas shorter RNAs are only weakly bound and unlikely to be cut. The simulations reveal how the exosome can act both as a molecular motor that pulls RNA, without energy input other than the one released in nucleotide cleavage, as well as an enzyme that cuts RNA. More information is available on our RNA exosome website.
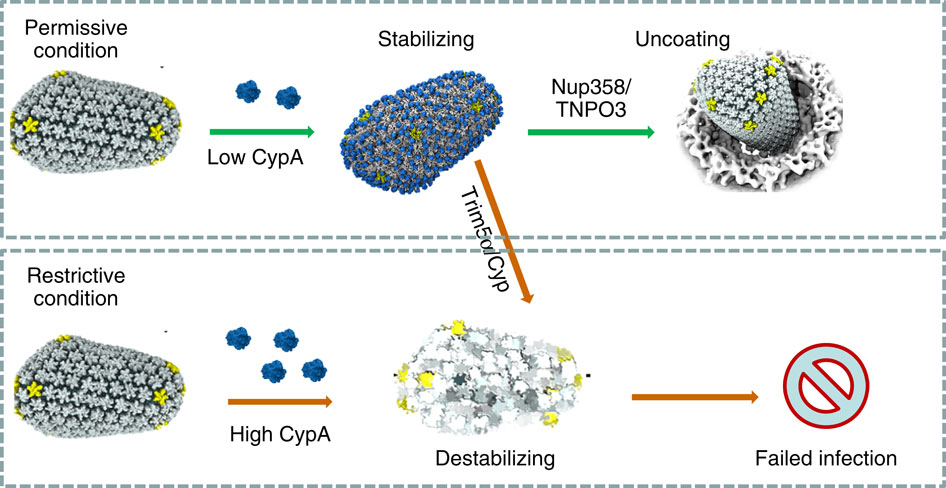
image size:
101.5KB
made with VMD
When experimental-computational biologists embarked on the great challenge of resolving the atomic level structure of the HIV virus capsid that contains the virus' deadly genetic cargo, they were advised by referees not to try as the capsid is too big, too irregular, and nobody would need the highly resolved structural information. Stubbornly, the researchers went ahead anyway and succeeded getting the atomic resolution structure, overcoming size and irregularity challenges (see highlight Elusive HIV-1 Capsid). But the question remained: Is the atomic level structure of the huge HIV capsid made of 1,300 proteins useless? The HIV capsid is a closed container made of protein pentamers and hexamers, with a surface of continuously changing curvature. Two recent experimental-computational studies demonstrate now that the capsid structure is far from useless, in fact, it is a great treasure. The first study was published last year and showed that the human protein, Cyclophilin-A (CypA), involved in several diseases, interacts with the HIV capsid and affects the capsid's dynamic properties (see highlight HIV, Cells and Deception). In a second, recent study, guided by cryo-EM measurements and benefiting from large-scale molecular dynamics simulations with NAMD, researchers could resolve with new accuracy the binding of hundreds of CypA proteins on the capsid's surface. They found that CypA binds along high curvature lines of the capsid, which enhances stiffness and stability of the capsid, even though only about half of the capsid is actually covered by CypA. The limited levels of CypA stabilize and protect the viral capsid as it moves through the infected cell towards the cell's nuclear pore where nuclear proteins additionally bind to the capsid at places not covered by CypA and promote there uncoating and release of the capsid cargo into the nucleus. More information is available on our retrovirus website and in a news release.
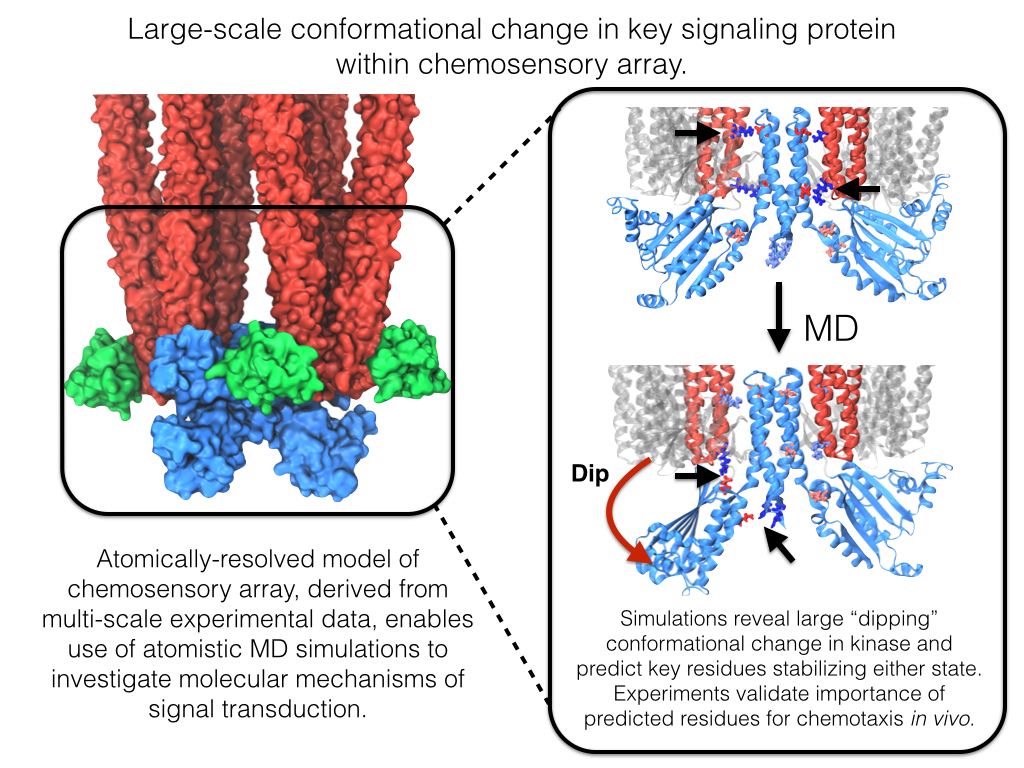
image size:
642.1KB
made with VMD
Motile bacteria position themselves within their habitats optimally, seeking proximity to favorable growth conditions while avoiding unfavorable ones. Cues used for this placement come in the form of small chemicals, so-called attractors and repellants, as well as physical factors such as favorable visible light and unfavorable UV radiation. To balance such a broad range of factors, bacteria monitor their environments and respond by way of a fundamental sensory capability known as chemotaxis. Chemotactic responses in bacteria involve large complexes of sensory proteins, known as chemosensory arrays, that process the information obtained from the bacteria's habitat to determine its swimming pattern. In this sense, the chemosensory array functions as a bacterial brain, transforming sensory input into motile output. Despite great strides in the understanding of how the chemosensory array's constituent proteins fit and work together, a high-resolution description of the kind needed to explore in detail the molecular mechanisms underlying sensory signal transduction within the array has remained elusive. A new study, utilizing cryo-electron microscopy and molecular dynamics simulations with NAMD, reports the highest resolution images yet of the bacterial brain's molecular anatomy. Using computational techniques, structural data from X-ray crystallography and electron microscopy are compared to derive an atomically resolved model of the chemosensory array's extended molecular structure that involves millions of atoms. Subsequent simulations of the model revealed a novel conformational change in a key sensory protein, that is interpreted as a key signaling event in the translation of chemosensory information into swimming pattern. More details on this work can be found in a recent news release as well as on our bacterial chemotaxis website.
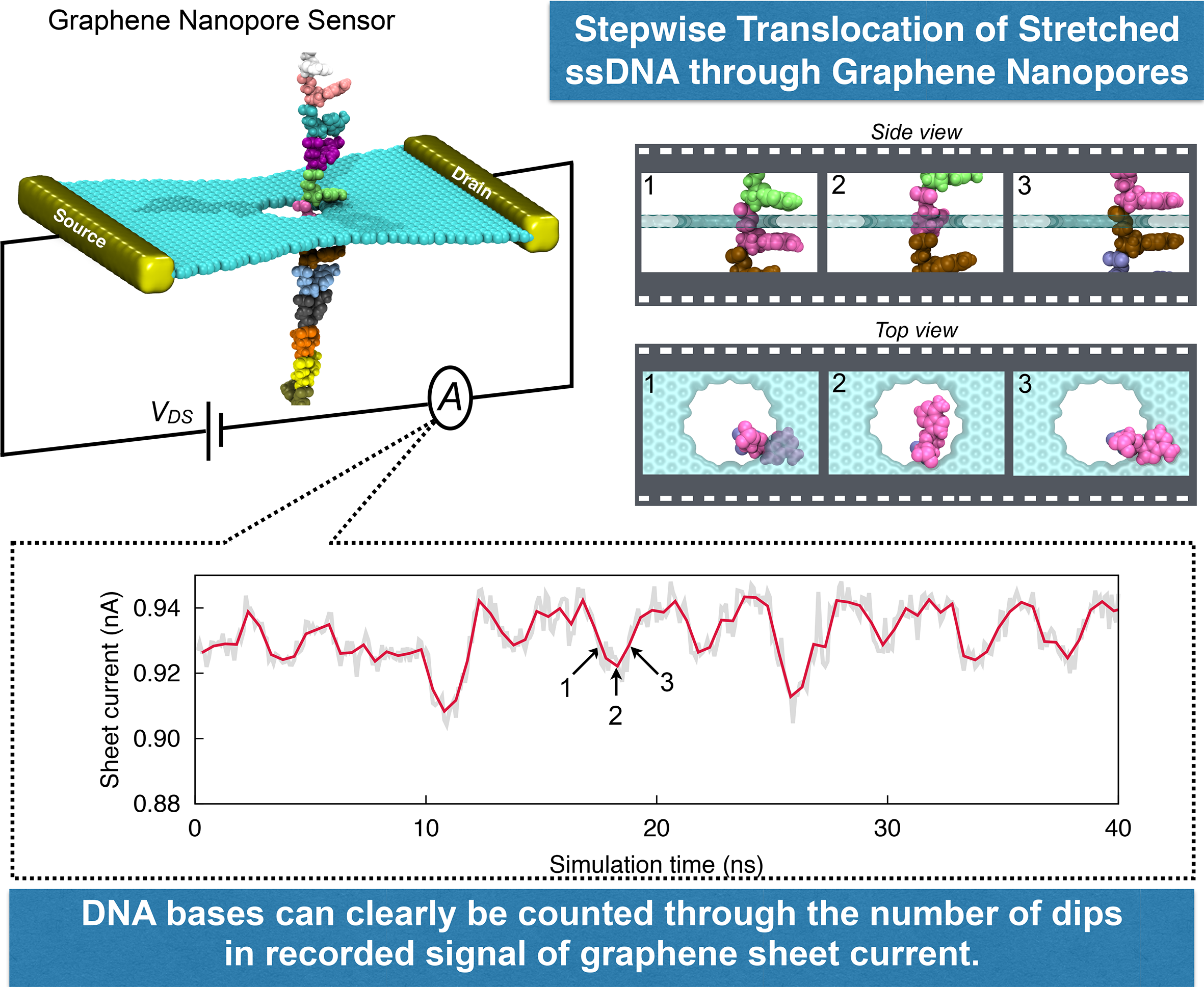
image size:
1.7MB
made with VMD
DNA sequencing is achieved by following a strand of DNA at a speed that permits recognition of the DNA bases in their actual order, thousands of bases or more for each pass. Nanotechnology can assist in the task, in principle, by furnishing sensors that can resolve single DNA bases and nano-mechanical actuators that pull the DNA in a controlled fashion passing through the sensor. Instead of building and testing actual sensors and actuators it is cheaper and faster to simulate them. Nanoengineers have indeed succeeded with such simulations over the last decade focussing on silicon technology (see October 2004 highlight: Transistor Meets DNA). Now the engineers have moved with their simulations to graphene technology that promises much better resolving power as sensors are thinner and as signals can be detected electronically in graphene (December 2013 and November 2014 highlights). The main unsolved problem is the mechanical actuator: how can one control movement of DNA through a graphene sensor such that measured signals become less noisy and bases can be recognized? A recent study, based on molecular dynamics simulations with NAMD and quantum electronics calculations of graphene, suggests use of an actuator that simultaneously stretches the DNA and pulls it through the sensor. This manipulation leads to a stepwise translocation of DNA through the graphene nanosensor, slowing down DNA translocation and stabilizing DNA bases inside the sensor. Read more on our graphene nanopore website.
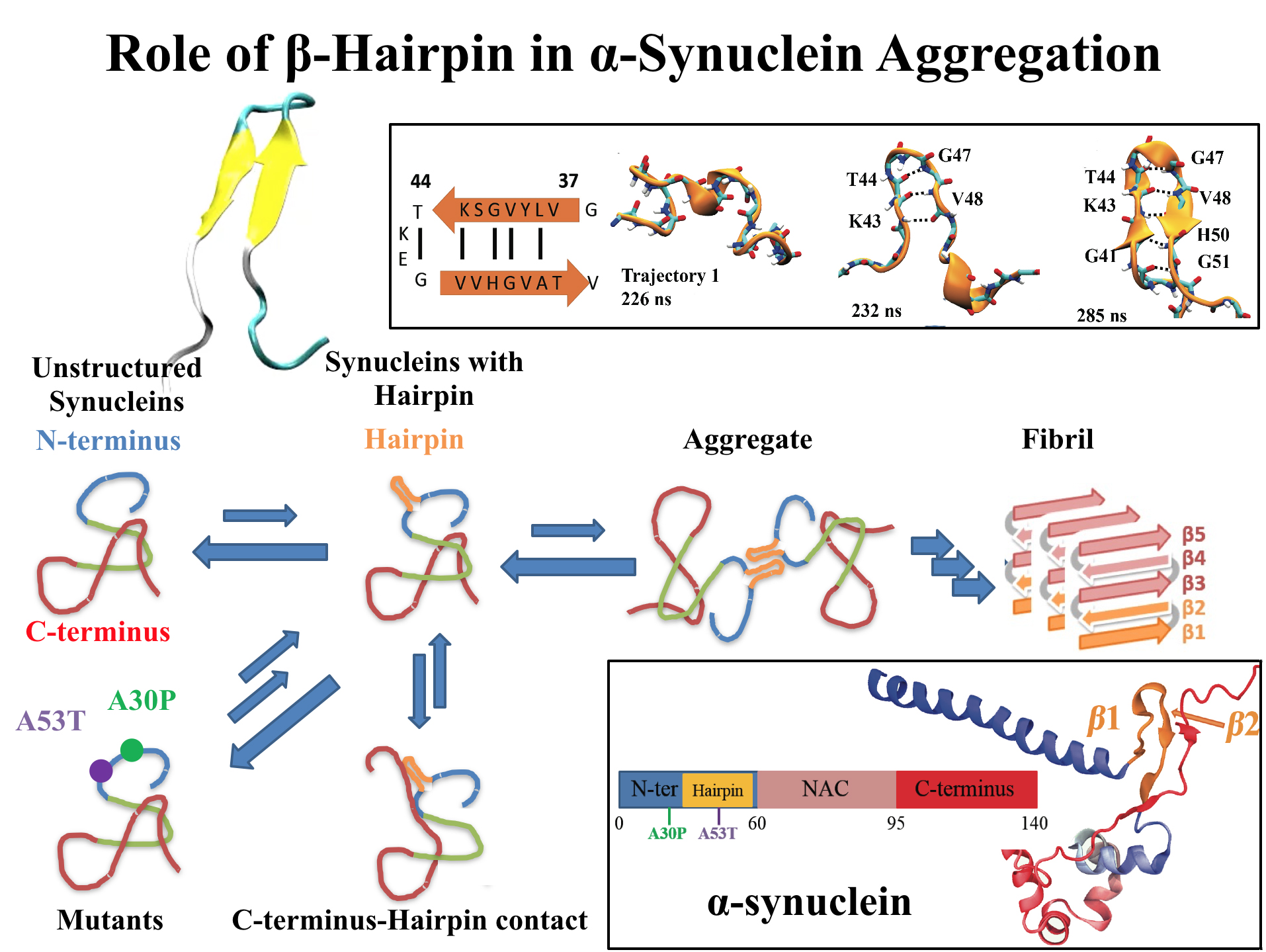
image size:
768.0KB
made with VMD
For centuries, millions of people around the globe have been troubled with a movement disorder that usually starts with a tremor in one hand. The disorder, later known as Parkinson's disease, affects commonly older individuals and disrupts patient's movement, sleep and speech from the brain. There is currently no cure for the disease. Key to the disease, progressively occurring in patient's brain, is the loss of neuron cells due to aggregation of a small protein named α-synuclein. Extensive studies have been carried out, yet the function of the protein remains a mystery. It is amazing that aggregation of such small proteins eventually leads to neuronal cell death and generates tremendous difficulties in peoples' life. In a recent report, a team of computational scientists attributed the cause of α-synuclein aggregation to a hairpin structure involving just a small region (amino acids 38-53) in the middle of the protein. With extensive simulations (over 180 μs in total), the researchers revealed that a short fragment encompassing region 38-53, exhibiting a high probability of forming a β-hairpin structure, is a key region during α-synuclein aggregation. Moreover, the researchers predicted a mutation that impedes β-hairpin formation, thereby retarding α-synuclein aggregation. The discoveries, made possible through the software NAMD and VMD, are expected to shed light on the mechanism underlying Parkinson's disease and to inspire the design of drugs. More on our α-synuclein website.
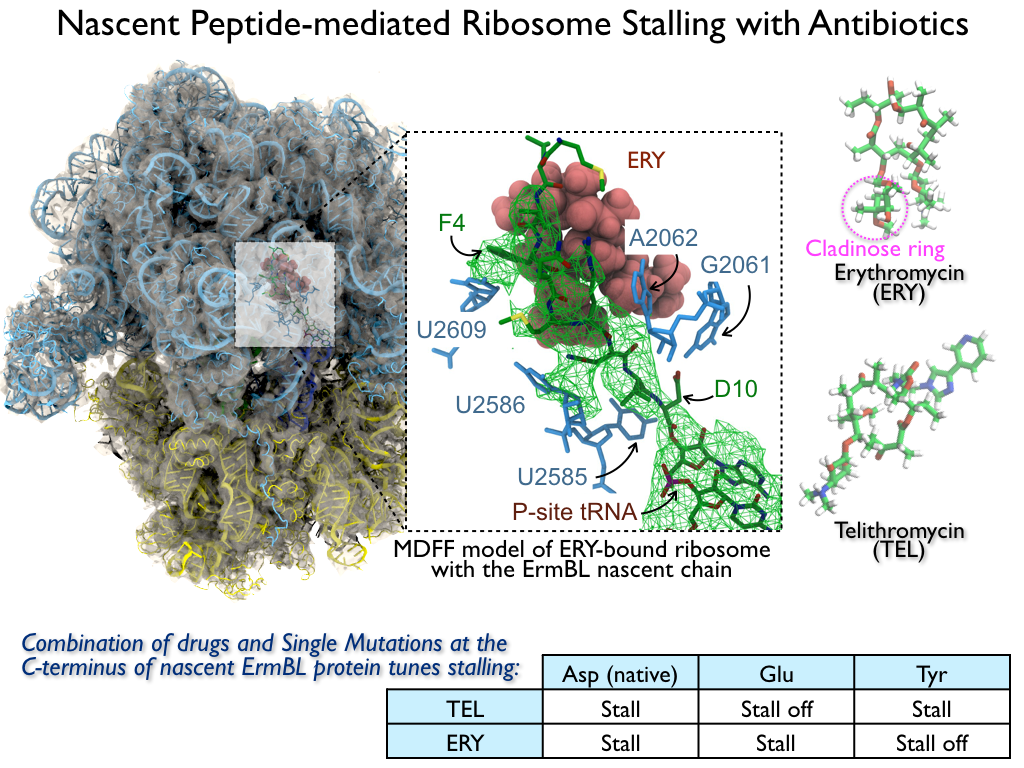
image size:
816.0KB
made with VMD
The ribosome is the ubiquitous machine in all living cells responsible for translating the cell's genes into functional proteins. The majority of antibiotic drugs target the ribosomes of bacterial cells while leaving human ribosomes unharmed. An example are the most widely-prescribed antibiotics, erythromycin and telithromycin. They kill bacteria by changing the properties of bacterial ribosomes and, thereby, disturb the bacterial protein production (see the Oct 2014 highlight Antibiotic Action on the Ribosome). However, modern bacteria fight antibiotic drugs; exposing them to a specific kind of antibiotic drug for too long will trigger the expression of drug-resistance genes, which protect the bacteria, eventually making the drug useless. Due to historical overuse of antibiotic drugs, clinic antibiotic drugs have experienced today serious drug-resistance problems. In a joint effort of computational and biomedical investigations, reported recently, molecular dynamics simulations with NAMD and systematic mutation experiments showed that the above antibiotics interact in a bacterial ribosome with a drug resistance gene - coded nascent protein and make it stall translation; however, engineered simple mutations in the bacterial gene can abolish stalling and, thereby, prevent the effect of drug resistance genes. The research suggests that engineered mutations might be a strategy to prevent antibiotic resistance. Read more on our Ribosome website.
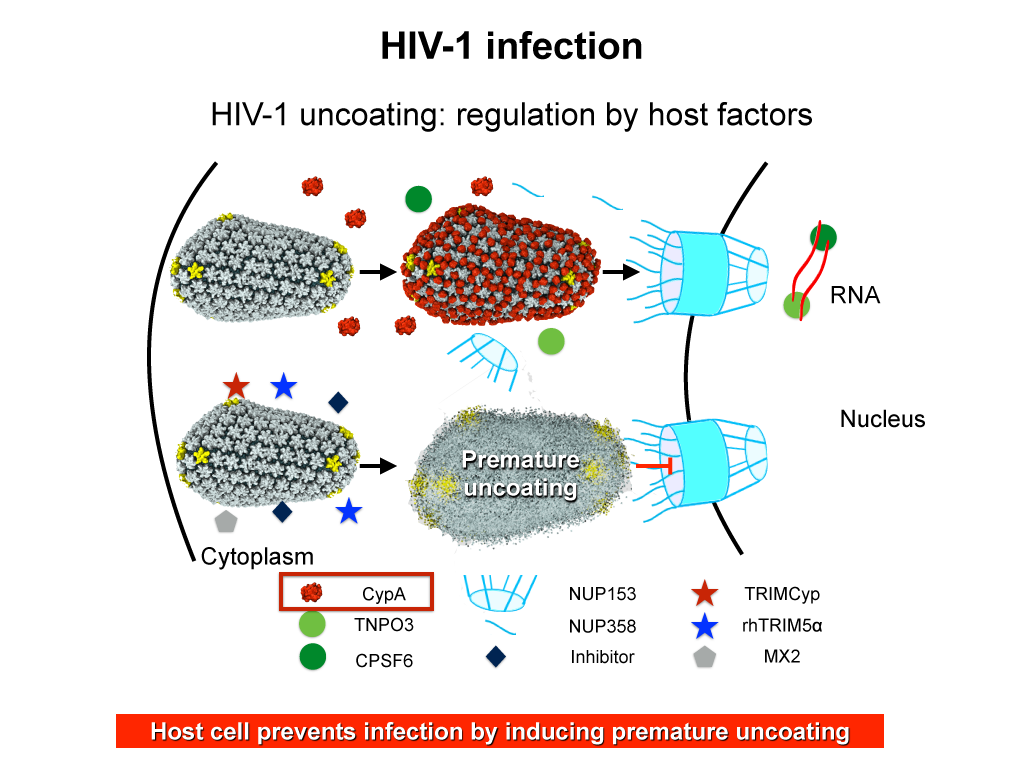
image size:
283.9KB
made with VMD
When human immundeficiency virus (HIV) infects a human cell, it releases into the interior of the cell its capsid (made of about 1,300 identical so-called CA proteins), a closed, stable container that protects the viral genetic material (see also June 2013 highlight Elusive HIV-1 Capsid and August 2015 highlight Anatomy of a Dormant Killer). Once in the cell ― while avoiding detection by cellular proteins ― the capsid deceives the cell and directs the cell machinery to transport it to the nucleus. The human-cell protein Cyclophilin A (CypA) is thereby exploited to act against the cell's well being and to assist the HIV infection by getting the capsid to access the cell nucleus; this results in a delicate choreography accomplished by escaping anti-viral proteins in the cell and deceiving transport proteins at the nucleus, all of which contain a CypA domain that interacts directly with the capsid. Despite the availability of the crystal structure of the complex of CypA and CA proteins determined nearly 20 years ago, the mechanism by which CypA assists the capsid has been unclear due to the lack of information on CypA in complex with not one CA protein, but the entire capsid. In collaboration with experimental groups, computational biologist have shown in a recent report that the effects of CypA on the capsid are not only structural, but also dynamical. Thus, new therapeutic strategies may be envisioned through modulation of the dynamics of the capsid by small-molecule (drug) compounds that inhibit the binding of CypA to the capsid. More information is available on our retrovirus website and in a YouTube video.

image size: 408.8KB
The Theoretical and Computational Biophysics Group (TCBG), funded by National Institutes of Health and the National Science Foundation, is not only the developer of the widely used computational biophysics programs NAMD and VMD, but is also an ardent teacher and trainer in computational biophysics. The group has just completed its 40th hands-on computational biophysics training workshop (see complete list), having now taught over 1,200 participants in intense, face-to-face, practical training sessions in small groups, typically of 30 students. Participants are faculty, postdocs, industry professionals, and graduate students. The training material, in the form of very extensive tutorials, is freely available on the TCBG tutorial website. The workshops are continuously improved, based on participant feedback and on the evolution of NAMD and VMD. TCBG intends to continue the workshops, and is presently developing a new generation of training features built directly into NAMD and VMD in the form of the new user interface qwikMD. For more information, see the TCBG training website.
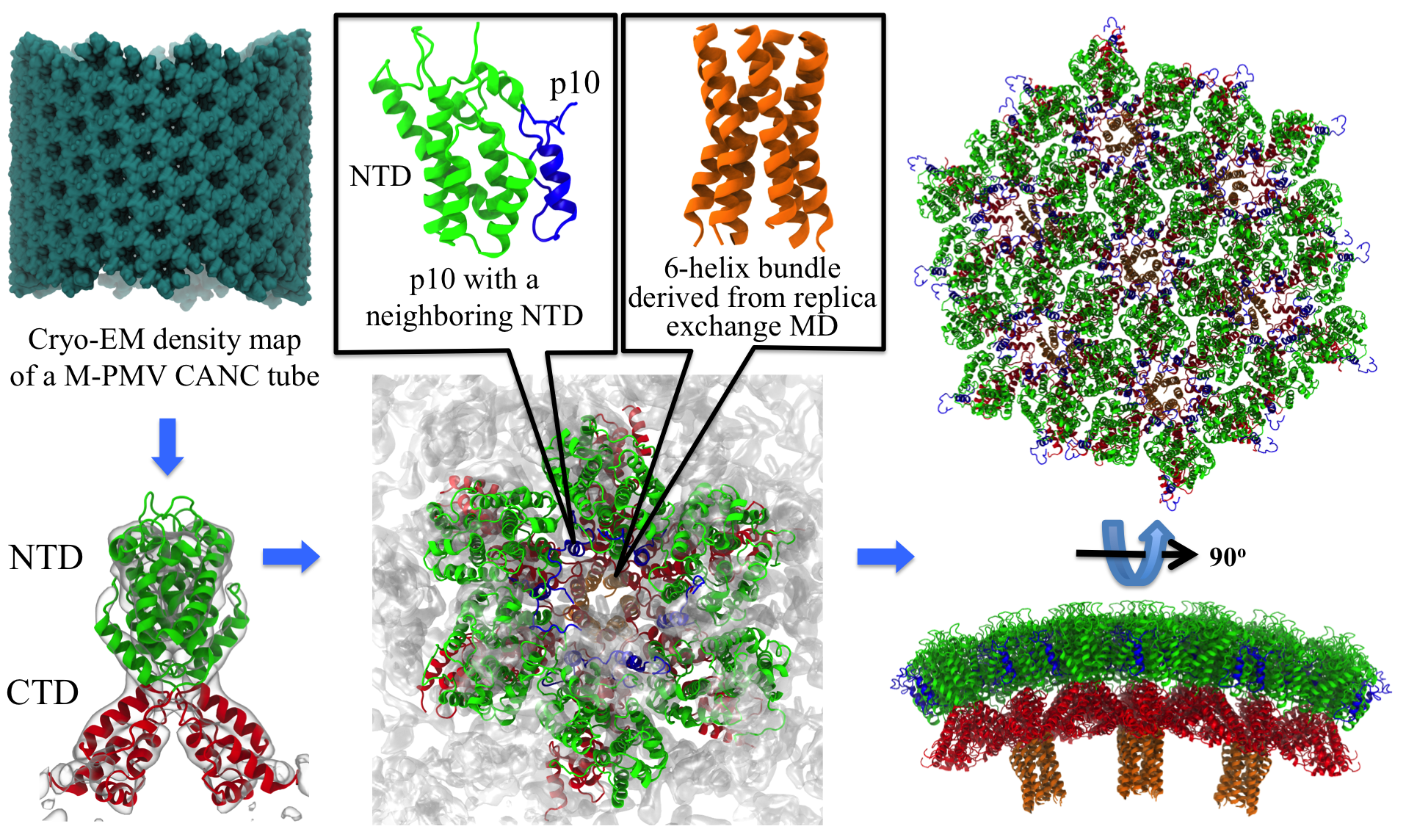
image size:
2.6MB
made with VMD
Retroviruses are parasites that pose a major health threat to humans (for example in case of HIV) and other animals (for example in case of RSV, M-PMV, MLV, and many more viruses). After a retrovirus hijacks a cell, the infected cell produces multiple copies of the virus which are then released into the host's bloodstream. These newly released viruses must mature before they can infect other cells. A strategy for preventing virus spread is therefore, to lock the viral particles in their immature, non-infectious state. However, to render the immature virus an attractive target for structure-based drug development one needs to know its chemical structure. Unfortunately, the complexity and size of the viral particle ― an incomplete hexagonal shell with a size close to 100 nm ― have prevented the experimental determination of the chemical, namely atomic level, structure of the virus. As reported recently, a team of computational and experimental researchers have provided an atomic structure of the immature retroviral lattice for the Rous Sarcoma Virus. The multi-domain RSV model was derived through a combination of state-of-the-art modeling techniques, including, cryo-EM-guided homology modeling, large-scale molecular dynamics simulations using enhanced sampling capabilities available in NAMD, together with experimental measurements such as X-ray crystallography and a wealth of biochemical data. Particularly, the model reveals novel features of the packing and dynamics of the immature capsid protein with implications for the maturation process and confirms the stabilizing roles of the so-called upstream and downstream domains of the immature RSV. More information is available on our retrovirus website, and in a highlight video.
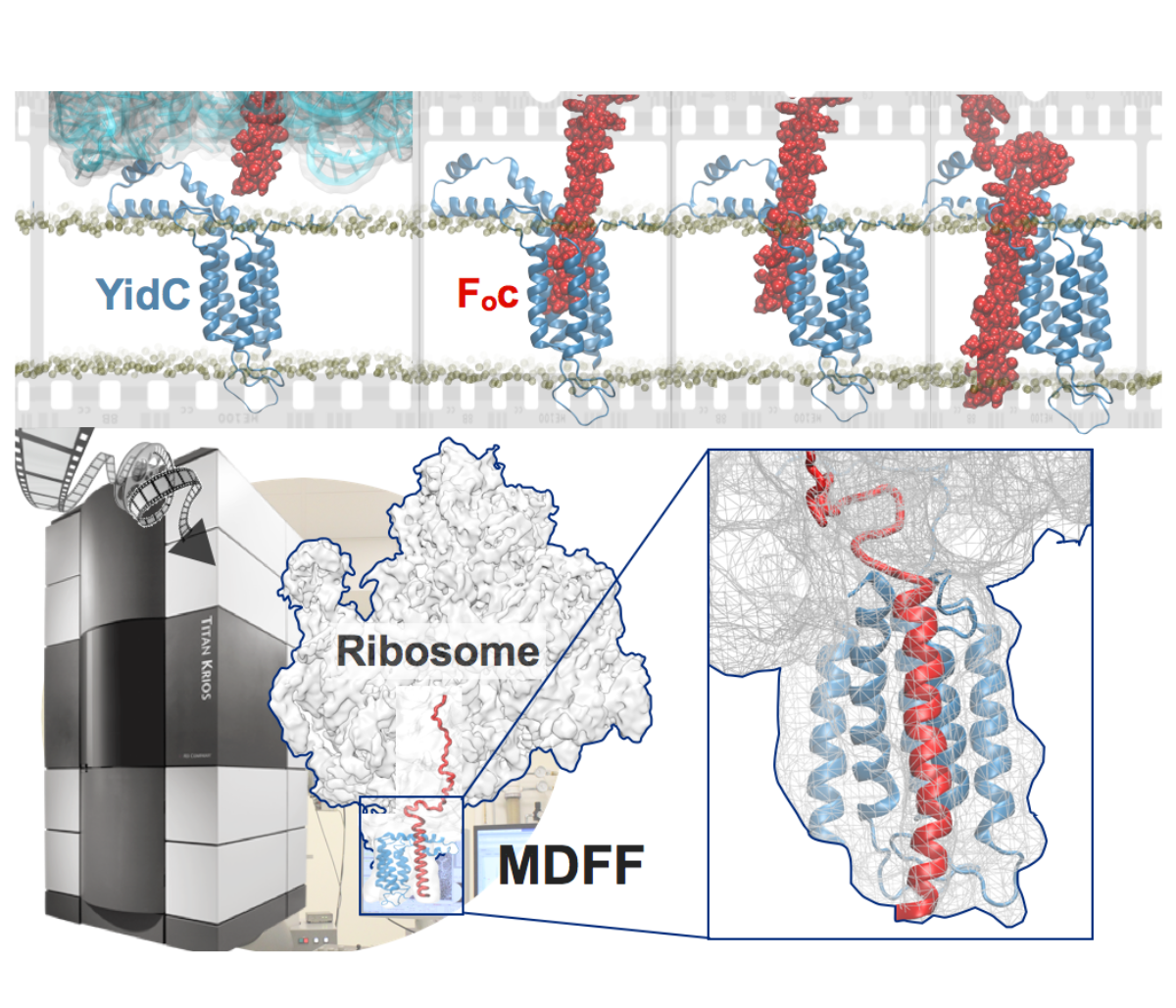
image size:
901.7KB
made with VMD
Synthesis and placement of new proteins in a living cell poses a challenge for the cellular machinery, in particular in case of so-called membrane proteins. Starting with nothing more than a sequence of DNA, the cell has to translate the genetic code, stitch together the constituent amino acids, and then place the newly made protein where its function is needed, namely the cell membrane. To meet the challenge the cell employs a molecular machine for the synthesis of proteins, the ribosome (see the Dec. 2009 highlight on Managing the Protein Assembly Line ), as well as special proteins that translocate newly synthesized proteins out of the machine into the cell membrane (see the Feb. 2011 highlight on Placing New Proteins ). Depending on the complexity of the membrane protein insertion, different protein systems are used for the translocation, in most cases the systems involving complexes of several, even many, proteins. Now however, the structure and function of the simplest translocating protein system has been solved, which actually is made of only a single protein, called YidC. Despite its simplicity, structure determination of YidC was difficult, taking three decades. Successful structure determination was recently reported here, and involved the combination of cryo-electron microscopy, mutational experiments and computer simulations, the latter using NAMD, VMD and MDFF. The structure that was discovered shows a distinctive arrangement of five trans-membrane helices and reveals how a single copy of YidC interacts with the ribosome at the ribosomal tunnel exit and identifies a site for membrane protein insertion at the YidC protein-membrane lipid interface. The quality of this atomic model is validated by its close agreement with a recently published crystal structure of E. Coli YidC (here). More on our protein translocation website.
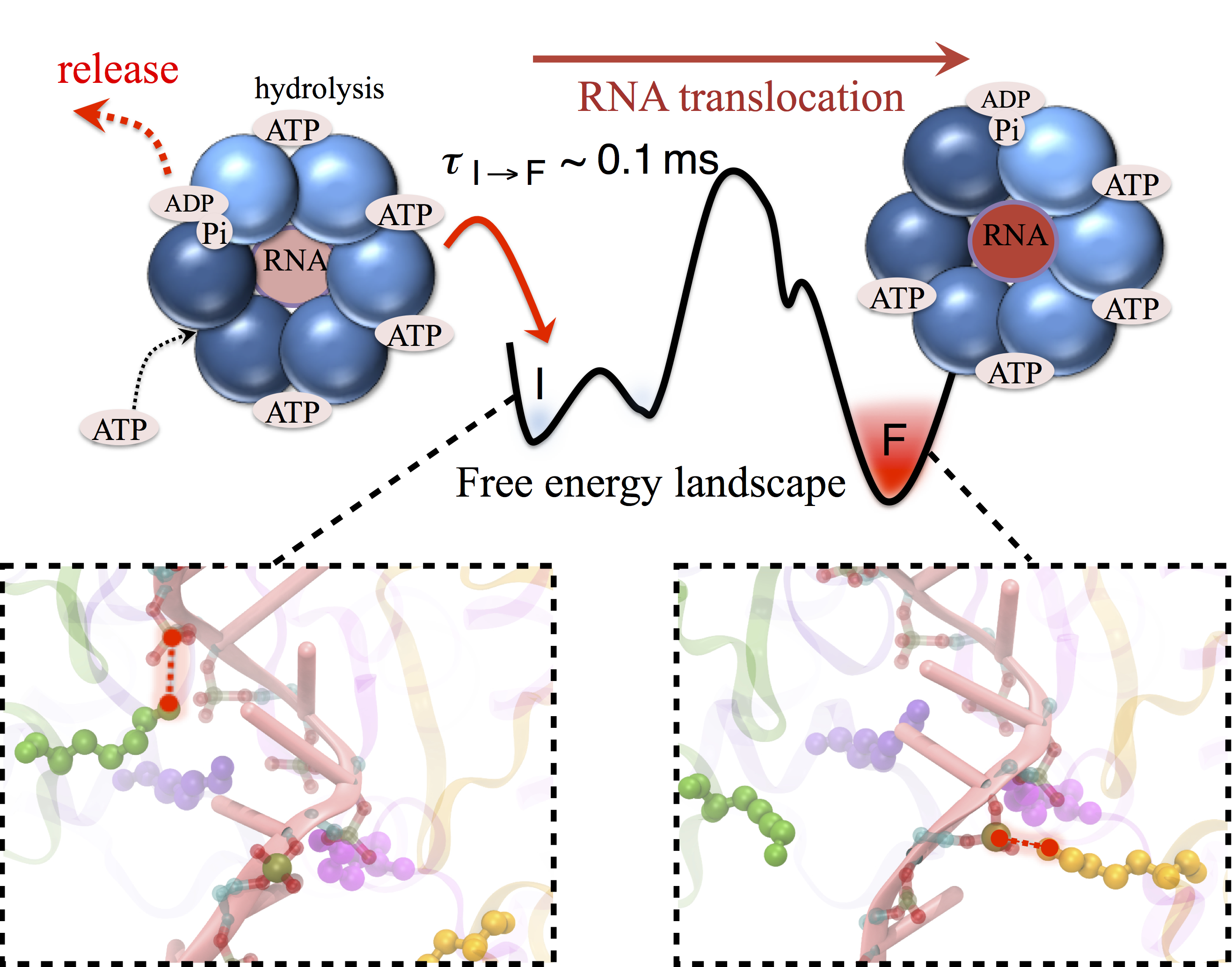
image size:
680.4KB
made with VMD
Many processes in living cells require molecular motors. Examples are transport of cargo within a cell, degrading misfolded proteins, and controlling gene expression. In the latter case acts a motor, called Rho, that moves along messenger RNA. The energy of the cell's motors stems from molecules of ATP that are converted to ADP, release thereby energy and drive motor action. How exactly this happens remained largely a mystery, despite decades of study and despite the availability of detailed molecular structures of the motors. A molecular dynamics study employing NAMD has achieved a great breakthrough in resolving the mechanism by which ATP-to-ADP conversion drives Rho to translocate along RNA. While molecular dynamics simulation, in principle, is well suited to explain Rho's motor action, the problem was that the action takes about a millisecond which is a time period beyond such simulations' reach. Employing new sampling methods, a recent publication, reported in new, complete and fascinating detail how Rho works. The simulations permitted literally to look under the hood of Rho's engine and see how it pulls itself along RNA and coordinates a cyclic and repeated motor action. It turned out that Rho, a ring of six identical protein subunits, engages in an ATP-to-ADP conversion-induced periodic motion of the subunits that pushes RNA electrostatically through the ring center. A completely surprising finding was the existence of coordination switches that make each ATP-to-ADP conversion lead to exactly one forward step along the RNA and keep the six subunits strictly synchronized, turning a randomly moving protein system into a well-behaved engine. More on our molecular motor website.
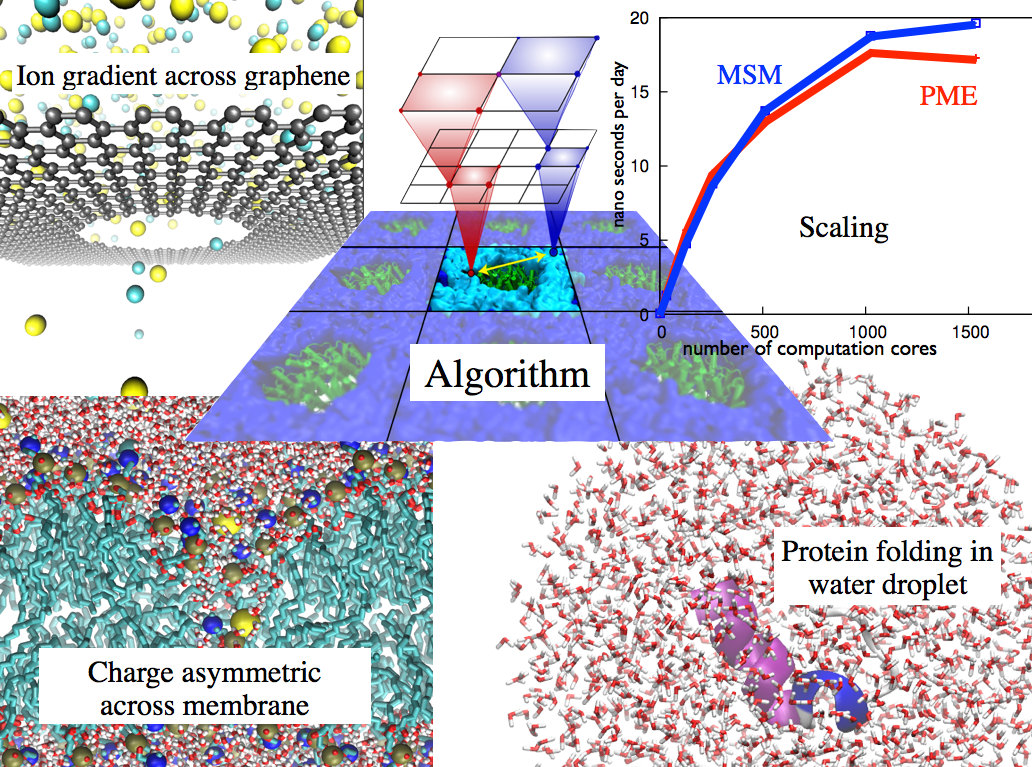
image size: 351.4KB
Long-range electrostatic interactions control macromolecular processes within living cells as prominent charges appear everywhere, such as in DNA or RNA, in membrane lipid head groups, and in ion channels. Reliable and efficient description of electrostatic interactions is crucial in molecular dynamics simulations of such processes. Recently a new mathematical approach for calculating electrostatic interactions, known as multilevel summation method (MSM), has been developed and programmed into NAMD 2.10 as reported here. Compared to the earlier decades-long approach, the particle-mesh Ewald (PME) method, MSM provides more flexibility as it permits non-periodic simulations like ones with asymmetric charge distributions across a membrane or of a water droplet with a protein folding inside. Furthermore, MSM is ideally suited for modern parallel computers, running, for example, simulations of large virus particles. More information here.
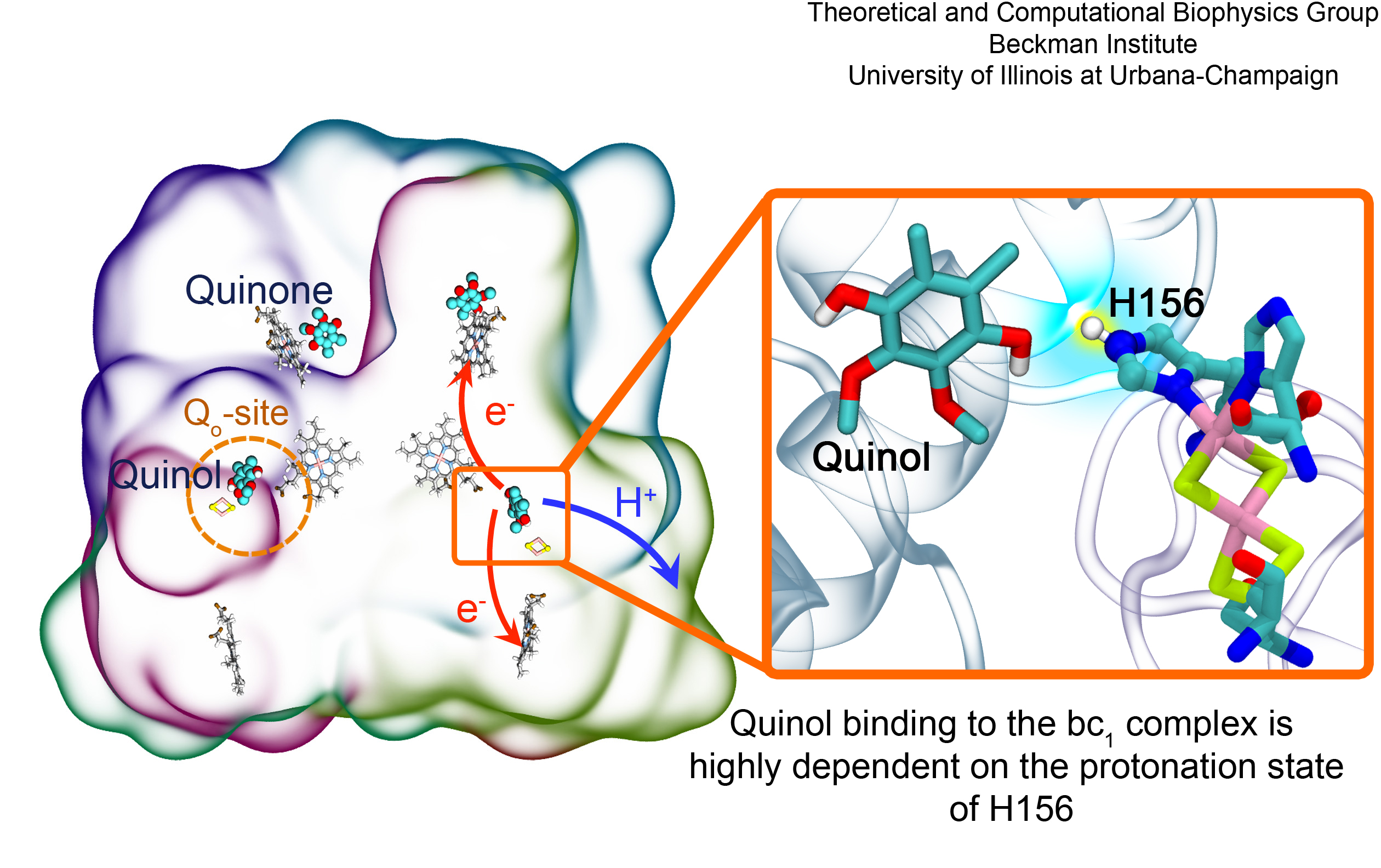
image size:
1.3MB
made with VMD
Most living cells acquire their energy through photosynthesis or respiration, both of which convert input energy (sun light or food, respectively) through coupled electron and proton transfer processes. A key role is played here by a protein, called the bc1 complex, that intermediately stores energy through the reaction of molecules of quinol into molecules of quinones, utilizing energy released to pump protons across an intracellular membrane. This reaction is initiated in the bc1 complex at the site of binding of the quinol molecule, but critical details about the physical mechanism leading to coupled electron-proton transfer are still unknown. A recent study, based on molecular modeling with NAMD and quantum chemistry calculations, investigated possible reaction mechanisms in case of the bc1 complex from the bacterium Rhodobacter capsulatus. The calculations suggest a novel configuration of amino acid residues responsible for quinol binding in the bc1 complex, and support a mechanism for coupled proton-electron transfer from quinol to iron-sulfur cluster. The study opens the door for a complete simulation description of the crucial role of the bc1 complex in bioenergetics. More about the bc1 complex can be found here.

The 1.9.2 release of VMD promises more insight and more beauty from use of an already powerful molecular visualization and analysis program. For more insight, VMD 1.9.2 exploits the power of parallel computers small and large to reduce analysis runtimes tremendously, as reported here, and here. VMD 1.9.2 strengthens collaboration between experimental and computational biologists in resolving atom-by-atom structure and dynamics of huge molecular assemblies arising in living cells by guiding interactively a match of computational model to experimental data, as reported here; this is achieved through quality-of-fit cross correlation to be computed rapidly using GPUs, the fastest means of modern calculation. Many new and updated tools, called plugins, developed by the VMD user community, are included in VMD 1.9.2, including force field parameterization, helix analysis, and normal mode plugins. VMD 1.9.2 incorporates a new remote control and works with Android phones and tablets. For more beauty, VMD 1.9.2 adds stunning interactive graphics on laptops. Such high quality graphics was previously available only on the most advanced computers, through powerful GPU-accelerated interactive ray tracing. Interactive ray tracing makes the task of getting a molecular image "just right" much easier than ever before; it also enables rendering of spectacular movies, turning scientists into great film directors. More details about VMD 1.9.2 features can be found here. See the light harvesting movie produced with VMD 1.9.2 here.
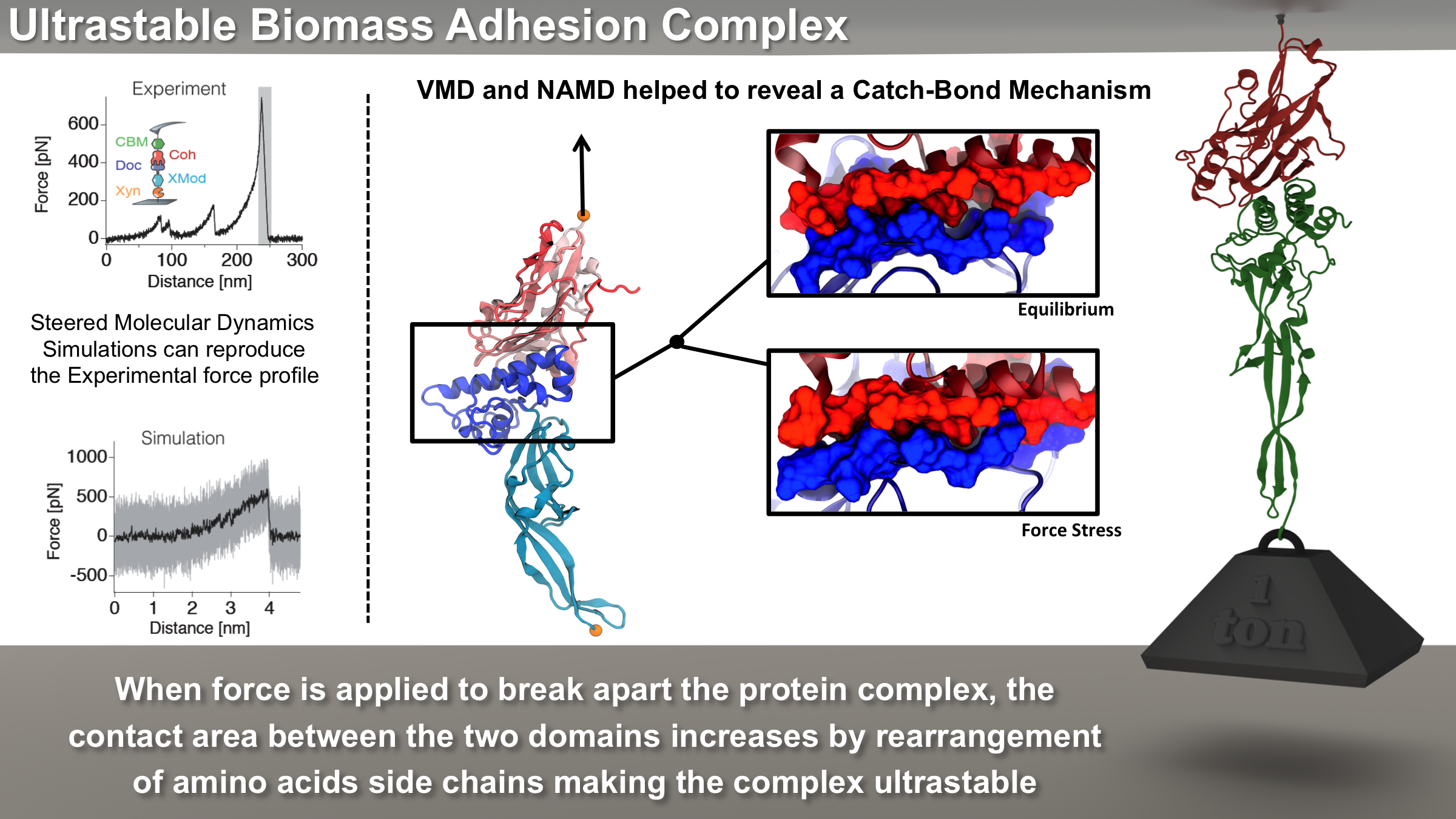
image size: 5.5MB
Bacteria can make a living from a very wide range of food sources. This ability makes them, for example, essential symbionts in animal digestive tracts where they assist their hosts in breaking cellulose fibers up into compounds degradable by the animal metabolism. Today, human gut bacteria, part of the human microbiome, are one of the hottest research topics in medicine. Gut bacteria face a particularly tough job in the rumen of the cow where they digest hardy cellulose fibers of grasses. Key to the job, taking place in a constantly moving fluid, are molecular tentacles, so-called cellulosomes, on the surface of the symbiotic bacteria. The cellulosomes develop a tight grasp on and then effective cleavage of cellulose. In a joint experimental-computational study researchers have investigated how in case of the bacterium Ruminococcus flavefaciens cellulosomes are built in a modular way, with molecular modules easily binding and unbinding during cellulosome construction, but sticking extremely strongly together during cellulosome digestive activity. As reported recently, single molecule force microscopy and molecular dynamics simulations using NAMD could show that under strain the adhesive bonds between cellulosome modules become stronger than seen in any other biomolecular system, in fact, become nearly as tight as strong chemical bonds. While the experimental data revealed bond strength and other characteristics, simulations reproducing the observed data provided a detailed view of the adhesive bond at atomic resolution, thereby revealing the physical mechanism underlying the uniquely adhesive property of cellulosomes. Gut bacteria and cellulosomes can be employed in 2nd generation biofuel generation (see highlight Waste into Fuel). More on gut bacteria and cellulosomes on our biofuels website.
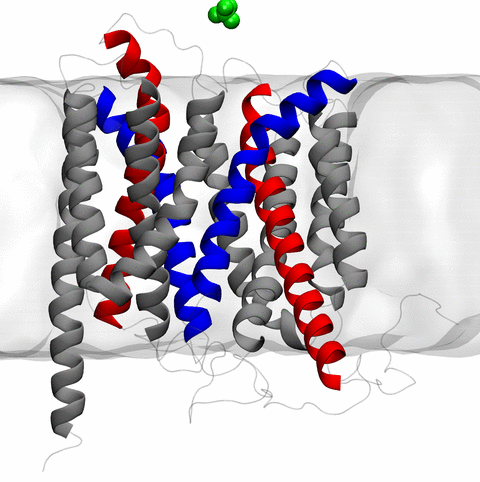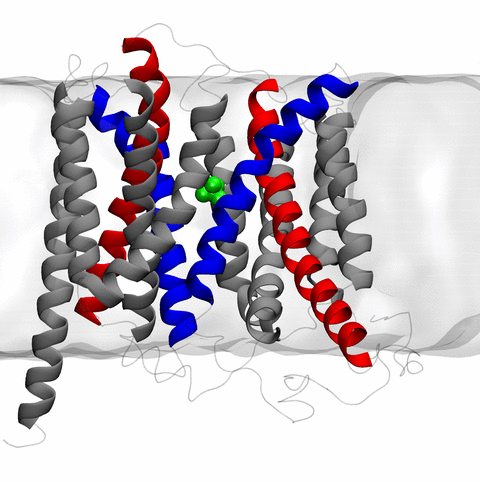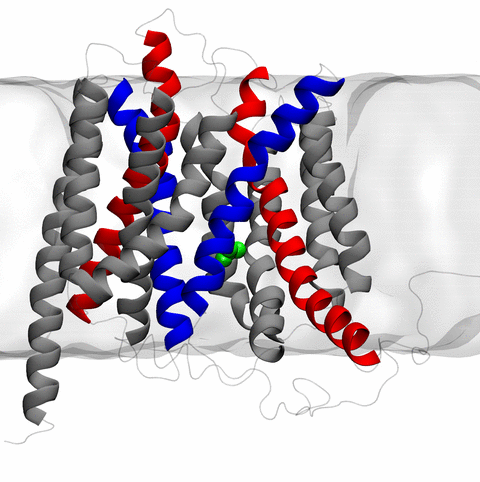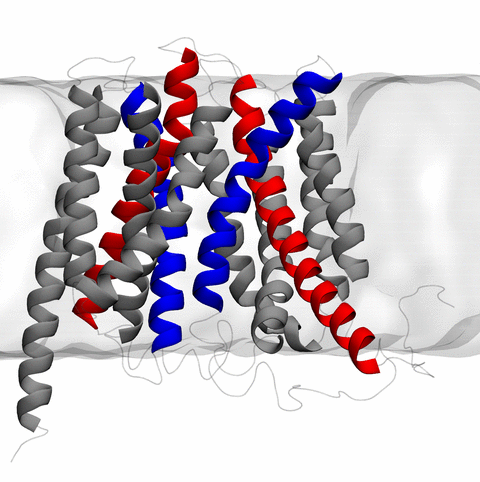
image size:
11.9MB
made with VMD
The 2.10 release of the molecular dynamics program NAMD includes numerous enhancements to support simulations of massive supramolecular assemblies such as the HIV capsid (see highlight Elusive HIV-1 Capsid). A single such simulation of a hundred million atoms or more can utilize the tens of thousands of processors of petascale supercomputers thanks to recent advances in computational methodology reported here. Equally if not more significant, however, are advances in NAMD's implementation of multiple copy algorithms for enhanced sampling of smaller molecular systems as reported here. These algorithms have already allowed researchers studying the molecular machinery of living cells to reveal for the first time with NAMD mechanisms operating on timescales of milliseconds or longer, including the rotary action of the ubiquitous energy conversion complex ATP synthase and the inward to outward opening transition of the membrane transporter protein GlpT, shown in the accompanying animation. NAMD 2.10 also introduces multilevel summation, reported here, a major algorithmic advance enabling efficient long-range electrostatics for non-periodic and semi-periodic simulations. More on new features in the 2.10 release of NAMD can be found here.
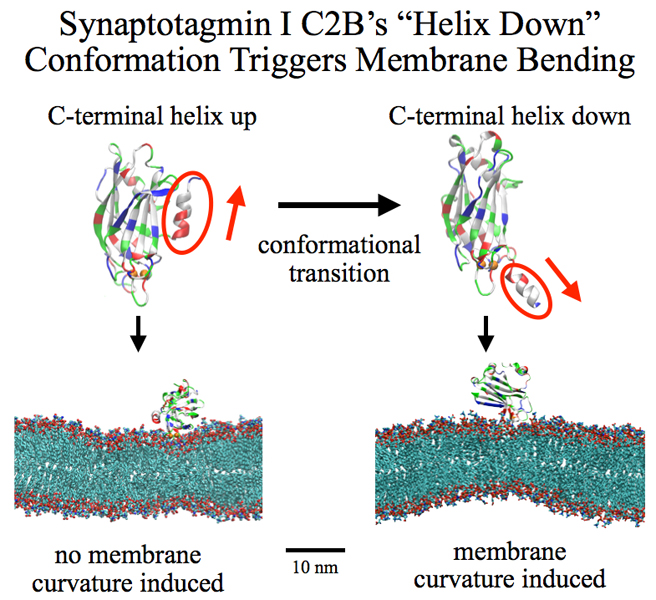
image size:
258.8KB
made with VMD
Neurons in the brain form a closely knit communication network with other neurons. Each neuron sends messages through up to thousands of cell-cell communication channels, so-called synapses. To avoid communication chaos, the messages of each neuron are chemically encoded as if neurons speak English to some neurons and French to others. The neurons employ an extremely efficient encoding system, packaging chemical message molecules, so-called neurotransmitters, in spherical vesicles encapsulated by a lipid membrane just like the whole neuron is encapsulated by a membrane. The vesicles aggregate near the presynaptic site of the membrane, ready to release their neurotransmitters into the space between neurons at the synapse, the so-called synaptic cleft. When a sender neuron becomes electrically active, as it wants to "speak", the electrical activity releases Calcium ions at the pre-synaptic cell that trigger merging (fusion) of vesicles with the sender neuron's membrane. At this point the neurotransmitter molecules flow into the synaptic cleft. The receiver neuron "hears" the signal by receiving the neurotransmitter molecules on receptors in the postsynaptic membrane, inducing as a result an electrical signal in the receiver neuron. The release involves a group of proteins that make vesicles ready for the release and proteins that execute the Calcium-triggered step, among the latter synaptotagmin I. As reported recently, researchers have proposed with the help of computer simulations using NAMD how synaptotagmin I acts. The simulations suggest that the experimentally observed structure of synaptotagmin I measured in vitro in a crystal/NMR form of the protein differs from the active, in situ form of synaptotagmin I. The finding, if true, will be a dramatic example for the role of computing in biology where the computer often complements observation studying, as in the present case, biomolecules in situ, namely their natural environment, rather than in vitro, namely in an artificial environment. Please read more on our neuron transmission website.

image size:
63.5KB
see also screencast
Who needs a supercomputer to do molecular dynamics? Thanks to Theodore Gray's new "Molecules App" for iPad/iPhone, you can now do molecular dynamics literally with the touch of a finger. "Molecules" by Theodore Gray is the interactive version of his beautifully illustrated book on the properties of the molecules that make up our world. Integrated into "Molecules" is the molecular dynamics code NAMD, which makes it possible to run real-time interactive simulations of the molecules in the book. You can pull, stretch, and twist hundreds of different molecules, or even tie them in knots! By playing with the interactive molecular dynamics simulations, readers can get an intuitive feel for the properties of molecules - not to mention, they are just plain fun to play with. The "Molecules App" is already very popular, and has been chosen for the Editors' Choice list on the App Store. You can see the app for yourself in the iTunes App Store, read more about the creation of the app on Theodore Gray's blog, and see a screencast of honorary group member Sebastian (pictured here) playing with NAMD through the app on the 500 atom molecule maitotoxin.
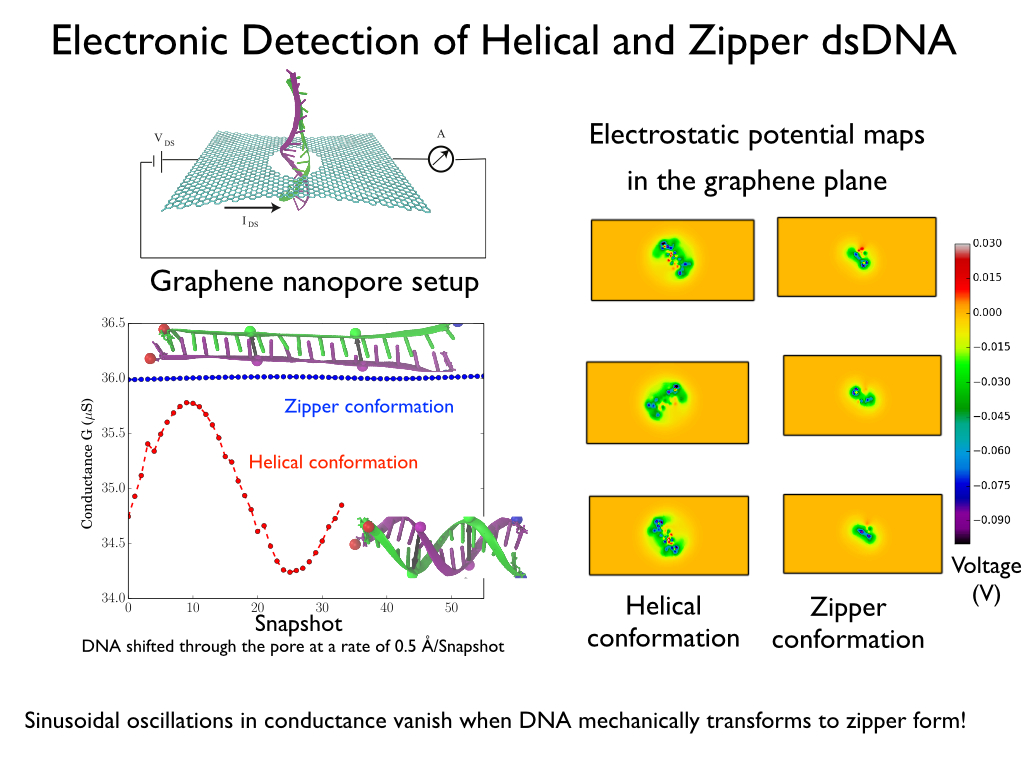
image size:
287.1KB
made with VMD
Threading DNA through a nanometer-size pore, so called nanopores, drilled into an ultrathin graphene membrane is a promising approach to build nanobiosensors for sequencing the human genome. Graphene nanopores can detect translocating DNA by recording concomitant flow of charged ions through the pore (see December 2011 highlight). As reported in the December 2013 highlight, graphene, which is an electrical conductor, offers a new way of sensing DNA molecules by monitoring sheet currents along the graphene membrane. DNA is a highly extensible molecule and upon mechanical manipulation can change its structure from a canonical helical conformation to a linear zipper-like conformation. A new study, which combines classical molecular dynamics simulations using NAMD with quantum mechanical simulations, suggests that sheet currents, in graphene membranes, can be used to detect conformation and sequence of a DNA molecule passing through the nanopore. This new research will guide the development of graphene-based nanosensors for DNA detection. More information can be found on our graphene nanopore website.
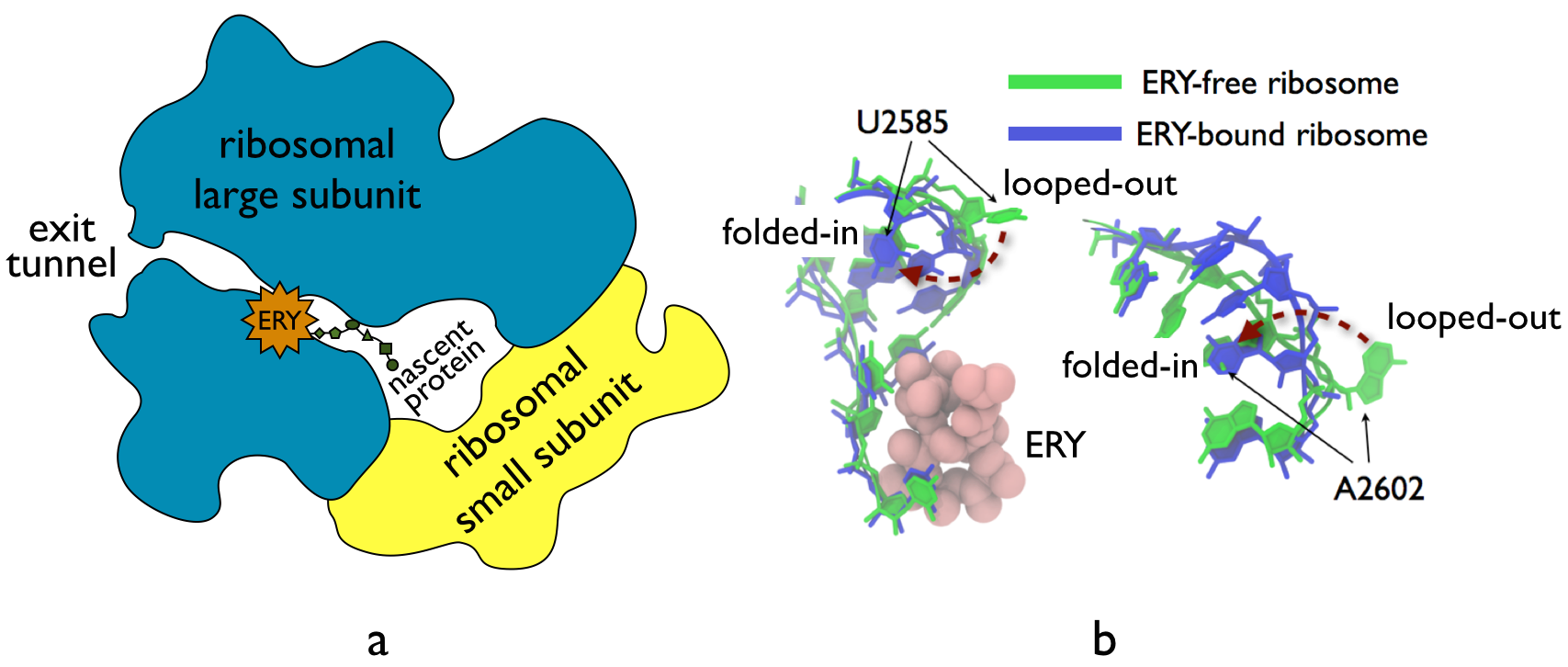
image size:
210.2KB
made with VMD
The ribosome, one of the ubiquitous molecular machines in living cells, is responsible for the critical task of translating the genetic code into functional proteins (See also Managing the Protein Assembly Line). The bacterial ribosome is the target of over 50% antibiotic drugs, for example, the clinically important macrolide family, including the widely-prescribed erythromycin (ERY) which is on the WHO essential medicines list. The antibiotic action of macrolide drugs has been known for over 50 years, however, the molecular mechanisms underlying the effects of these drugs are still unknown. It was previously believed that the antibiotic action by macrolide drugs has to be assisted by the presence of a nascent protein inside the ribosome. However, in a recent study, computational investigations jointly with biochemical experiments have revealed that the macrolide drugs can take an antibiotic action by altering the structure of the bacterial ribosome before translation of nascent protein really begins. Please see more highlights on translational control of the ribosome: Born to Control, Shutting Down the Protein Factory. Read more on our Ribosome website.
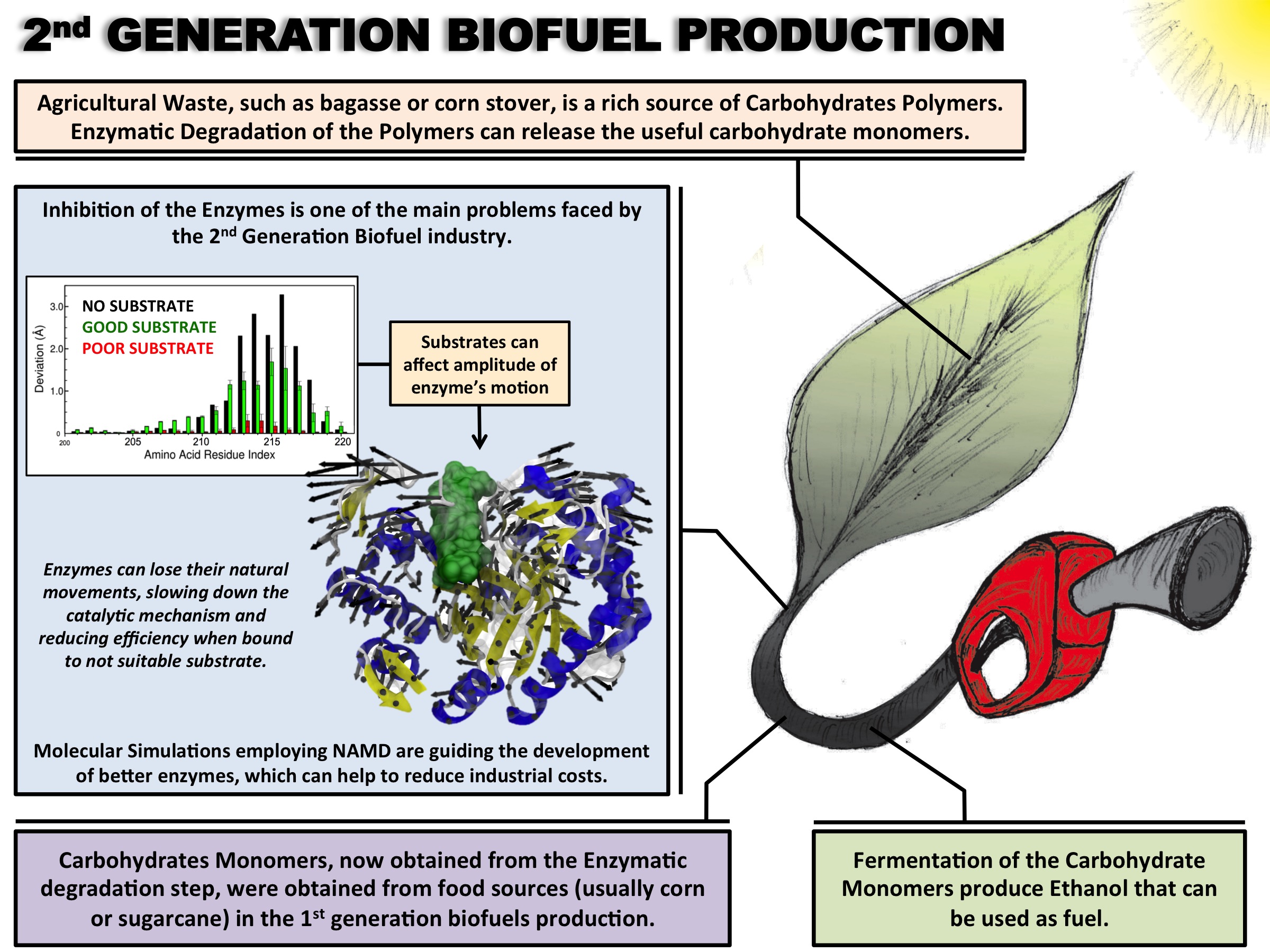
image size: 620.6KB
Biofuels are a well-known alternative to the largely used fossil-derived fuels, however the competition with food production is an ethical dilemma. Fortunately a solution is offered by second-generation biofuels, which can be produced from agricultural waste, or more specifically, from plant cell wall polysaccharides. Using the strategy of microorganisms, several enzymes are employed in the production of this advanced biofuel. However the biofuel industry faces problems such as the loss of efficiency of the enzymes that arises over time due to intermittent high concentration of non-suitable substrates. Simulations can guide biochemical experiments aimed at investigating the mechanism that makes the enzymes vulnerable to such substrates, helping the development of more efficient and, thereby, less costly enzymes. A recent study, based on molecular modeling with NAMD, reported that reduction of efficiency in an important enzyme, know as Man5B, is associated with a loss of the enzyme's flexibility. Molecular dynamis simulations showed that a poor substrate slows down a crucial opening and closing movement of the enzyme's catalytic cleft while a good substrate keeps the movement almost intact. The insight is of crucial importance since it suggests mutations to enzymes presently employed in second-generation biofuel production that need to be replaced less often and, thereby, rendering the production more cost-effective. Read more on our biofuels website.
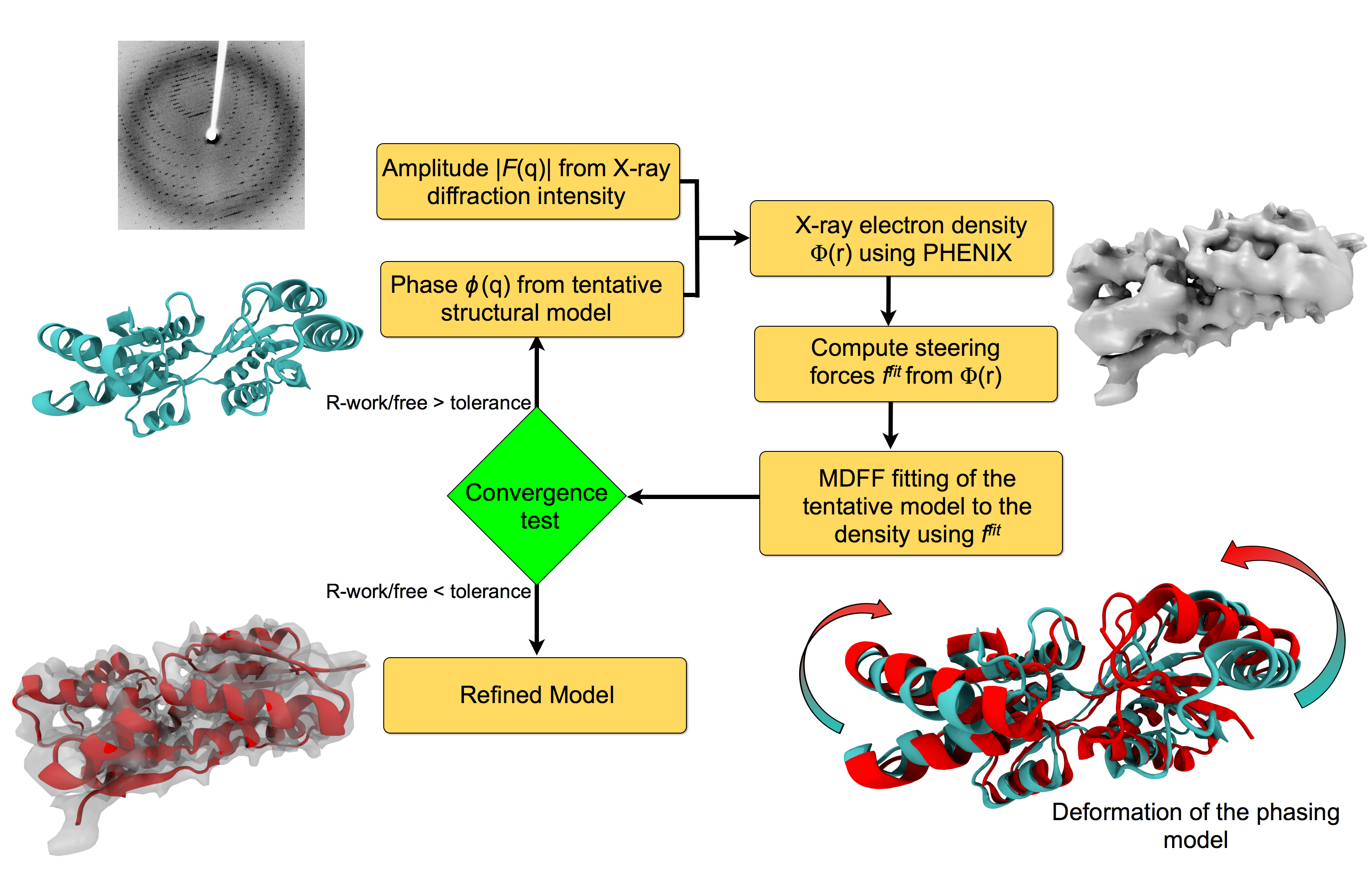
image size:
151.9KB
made with VMD
For many, the word 'X-ray' conjures up the images of white bones on black backgrounds hanging on the wall of a doctor's office. However, X-rays have played another important role for the past 100 years through their use in the determination of chemical structures at atomic level detail, starting with the first ever structure of table salt in 1924. Since then, the diffraction properties of X-rays, when shone on a crystal, have been used to solve increasingly large and complex structures including those of biological macromolecules found inside living cells. X-ray crystallography has become the most versatile and dominant technique for determining atomic structures of biomolecules, but despite its strengths, X-ray crystallography struggles in the case of large or flexible structures as well as in the case of membrane proteins, either of which diffract only at low resolutions. Because solving structures from low-resolution data is a difficult, time-consuming process, such data sets are often discarded. To face the challenges posed by low-resolution, new methods, such as xMDFF (Molecular Dynamics Flexible Fitting for X-ray Crystallography) described here, are being developed. xMDFF extends the popular MDFF software originally created for determining atomic-resolution structures from cryo-electron microscopy density maps (see the previous highlights Seeing Molecular Machines in Action, Open Sesame, Placing New Proteins, and Elusive HIV-1 Capsid). xMDFF provides a relatively easy solution to the difficult process of refining structures from low-resolution data. The method has been successfully applied to experimental data as described in a recent article where xMDFF refinement is explained in detail and its use is demonstrated. Together with electrophysiology experiments, xMDFF was also used to validate the first all-atom structure of the voltage sensing protein Ci-VSP, as also recently reported. More on our MDFF website.
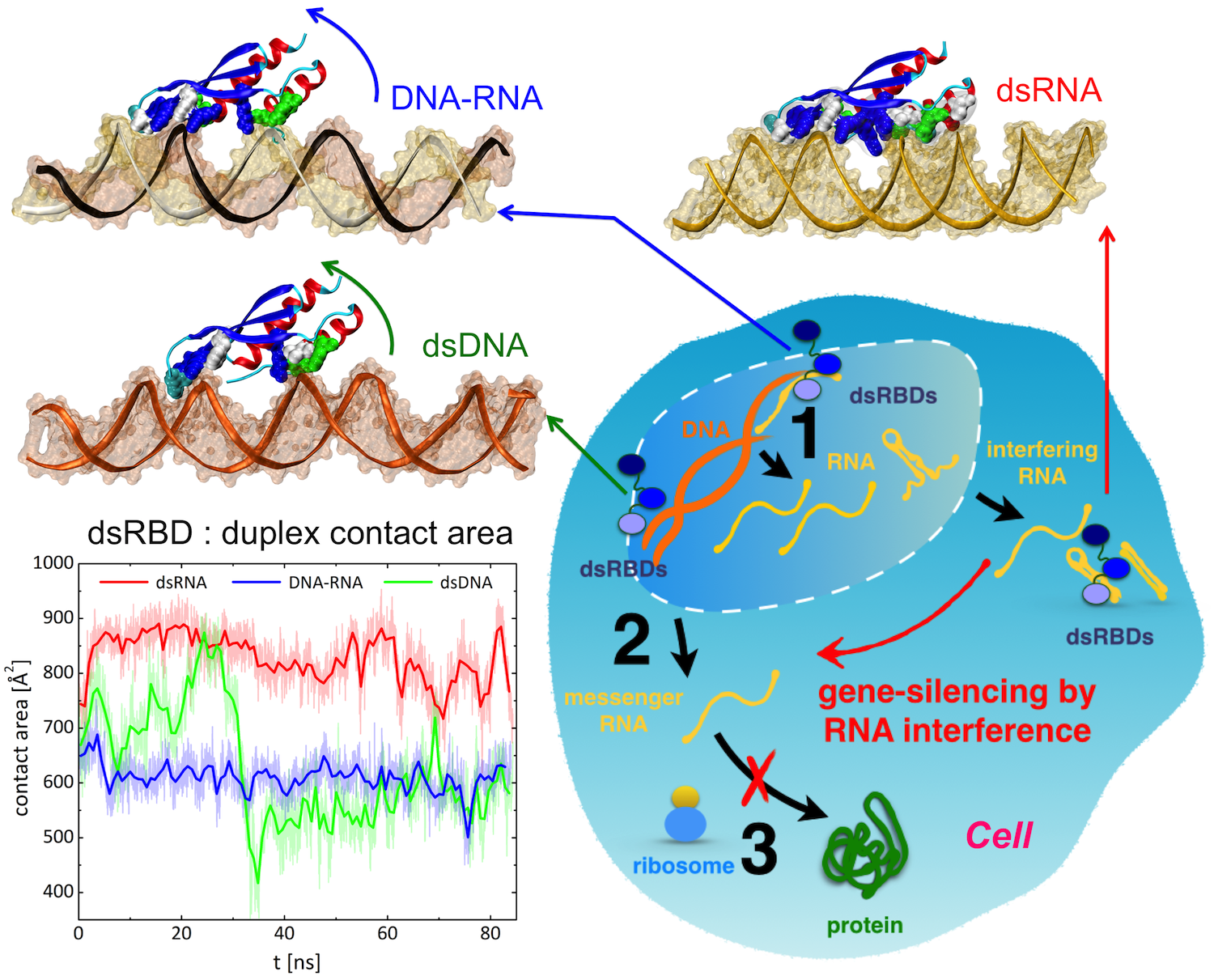
image size:
1.6MB
made with VMD
Only 2% of human DNA involves genes that code for proteins, the machinery of our cells. The DNA with genes is copied into mRNA read by ribosomes that synthesize then the respective proteins. However, 80% of the remainder DNA is also copied into RNA molecules that assume then manifold functions, one being that the RNA molecules bind to mRNA and silence thereby the respective genes. Frequently, gene-silencing RNA is activated with the help of proteins called double stranded RNA binding domains (dsRBDs). These domains bind to parts of gene-silencing RNA that happens to form double strands, similar but not identical to the double strands formed by DNA and discovered long ago by Watson and Crick. In fact, there exist small but characteristic differences between double stranded RNA, double stranded DNA and hybrid double strands made of RNA and DNA. In a recent study, computational biologists performed simulations using the molecular dynamics program NAMD to determine how dsRBDs recognize silencing RNA and discriminate between double stranded RNA, DNA, and hybrid double strands. The simulations revealed that dsRBDs and double stranded RNA fit together ideally like matching pieces of a puzzle, with mutually compatible shapes and electrostatic patterns. On the other hand, dsRBDs and double stranded DNA or hybrid double strands have poor fits due to changed and insufficiently flexible double strand forms. More here.
Atomic force microscopy (AFM) gives us a low-resolution glimpse of life at the nanometer scale. Now scientists can bring microscopy images to life by combining the microscopy data with atomic-detail structures to re-create the imaged system on the computer. As recently reported, Center scientists constructed an atomic-resolution model of a photosynthetic membrane based on AFM data showing the locations of the many light-harvesting proteins that inhabit the membrane. After using NAMD on petascale computers, like Blue Waters and Titan, to relax the 20-million atom membrane, Center scientists used the model to study the migration of energy among the light-harvesting complexes, as well as the mobility of quinone molecules in the membrane. The photosynthetic membrane patch was found to have a very high (90%) light-harvesting efficiency; further, it was found that the light-harvesting proteins could be considerably less tightly packed in the membrane with minimal loss of efficiency. Read more on our website.
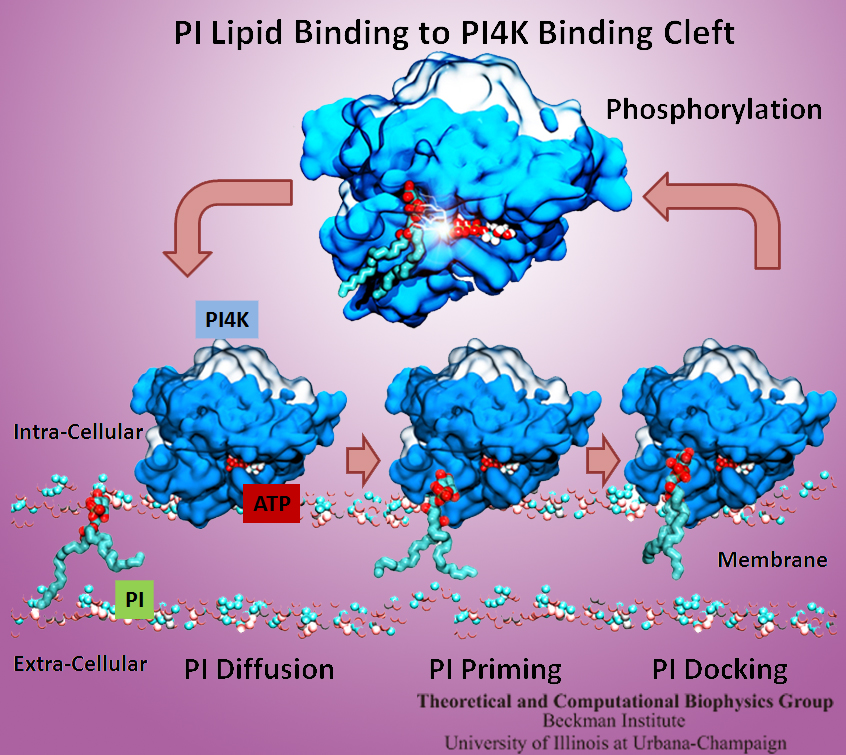
The Golgi apparatus found in so-called eukaryotic cells acts like Amazon.com, namely accepting delivery of newly synthesized proteins, packaging them, and sending them out. However, in comparison to Amazon.com the Golgi apparatus uses immensely more advanced packaging materials made of a multitude of lipids. The various lipids form membranes in the shape of vesicles. Depending on lipid type specific goods are packaged inside the vesicles, specific locations in the cell receive the packages, content is emptied there and packages are retrieved. To achieve the series of steps just outlined, lipids as the main actors need to be coordinated. One way is to recognize lipids forming vesicle membranes and to modify them to be readied for a subsequent step, for example going from release step to retrieval step. For this purpose eukaryotic cells engage a special class of proteins, named kinases, that can recognize membrane lipids and phosphorylate them, adding a so-called phosphate group. As reported recently, a team of experimental and computational scientists determined the atomic structure of a key member in the kinase family, phosphatidylinositol 4-kinase (PI4K). The scientists discovered not only the structure, but also how PI4K captures and phosphorylates a particular type of lipid molecule, thereby changing a vesicular membrane and turning on the next step in the cellular package delivery system. The discoveries, made possible through the software NAMD and VMD, are expected to have an impact on the design of novel drugs that suppress cancer cell growth. More on our kinase website.
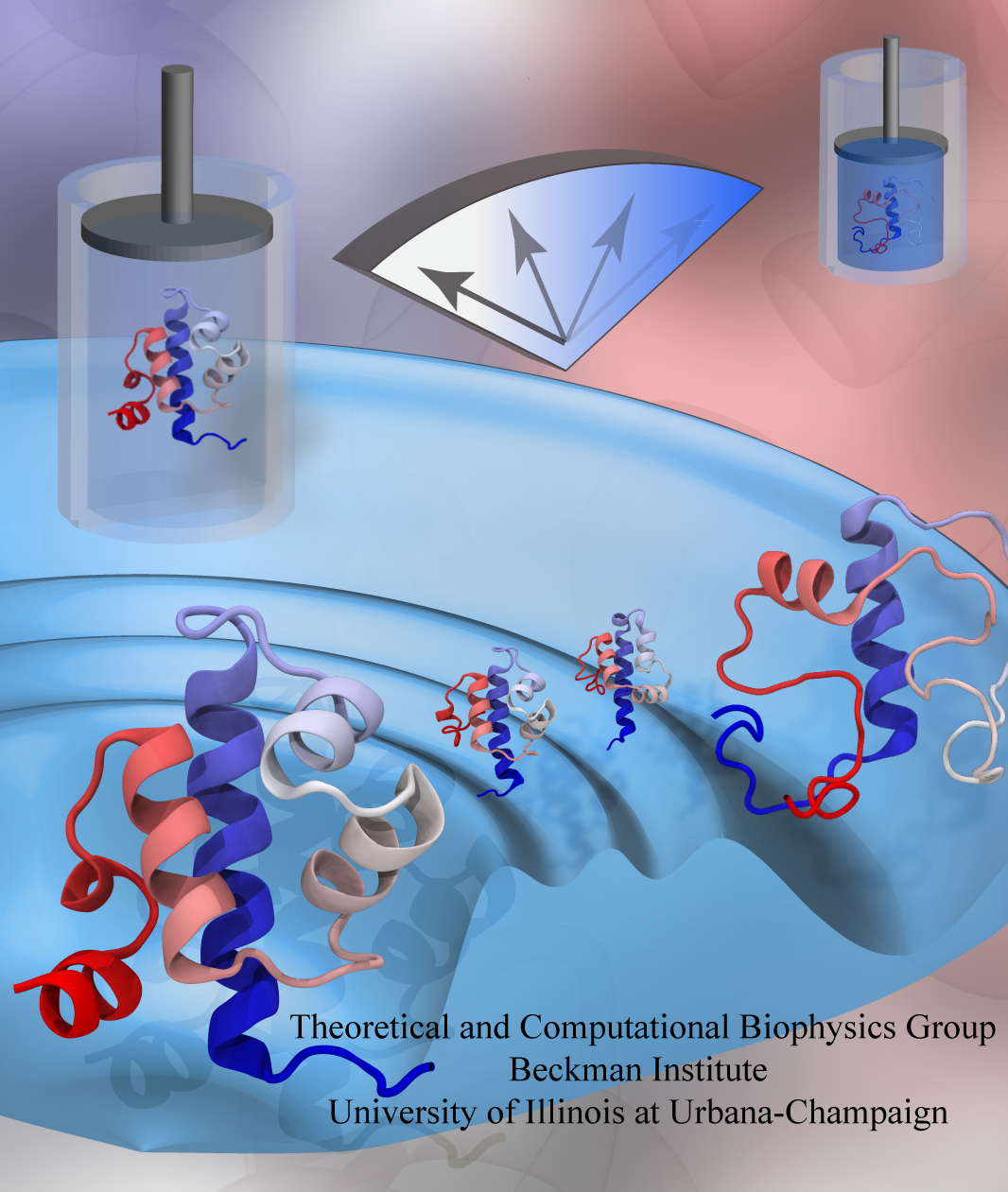
While pressure can help in cooking your favorite meat for dinner, pressure is also helping scientists to study how proteins, a key ingredient in any meal, loose and regain their proper shape. Proteins are key building blocks for any life form on earth, making the many machines that drive living cells. For any protein to do its job correctly, it has to first settle into the proper shape, the so-called native state. The process, referred to as protein folding, is still a mystery (see July 2012, Nov 2009 and May 2008 highlights). A general consensus is that the mystery can be solved only through a combination of experimental observation and computer simulation. In two recent reports (1 and 2), a team of experimental and computational scientists have used high pressure to force a protein to loose its proper shape, similar to what happens in a pressure cooker. After the high pressure is released in the experiment, the protein regains its proper shape, apparently by following two folding pathways, one fast and one slow. Using the molecular dynamics simulation program NAMD, as well as a special purpose supercomputer, Anton, the researchers were able to identify these two pathways and to follow every single folding step of the protein at an unprecedented precision. The studies greatly improve scientific understanding at the molecular-level of how proteins respond to pressure changes and, while not giving delicious recipes for pressure cooking in the kitchen, they are likely to suggest how to use pressure to dissolve toxic proteins that arise in disease, such as in Alzheimer's disease. More on our protein folding website.
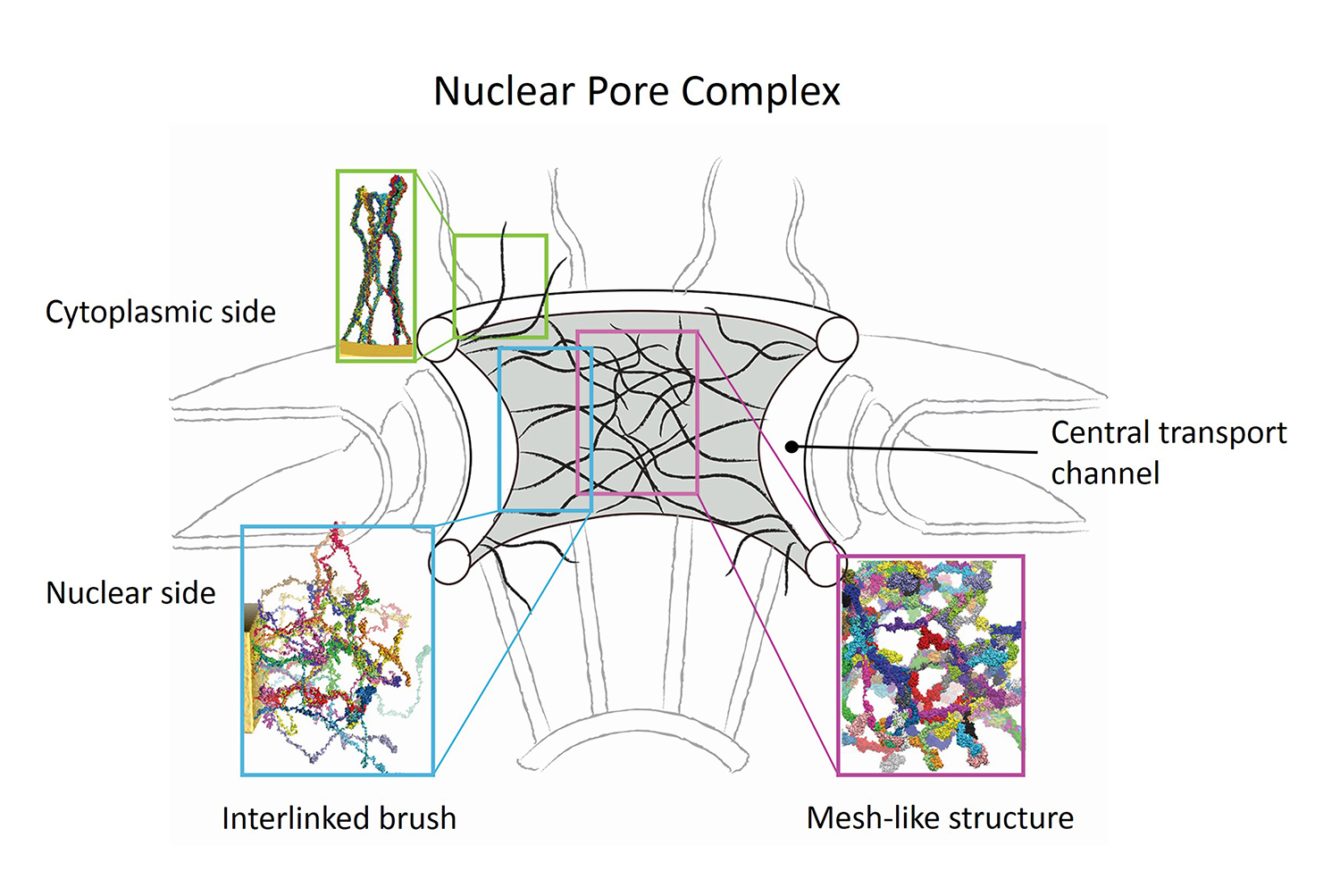
image size:
579.0KB
made with VMD
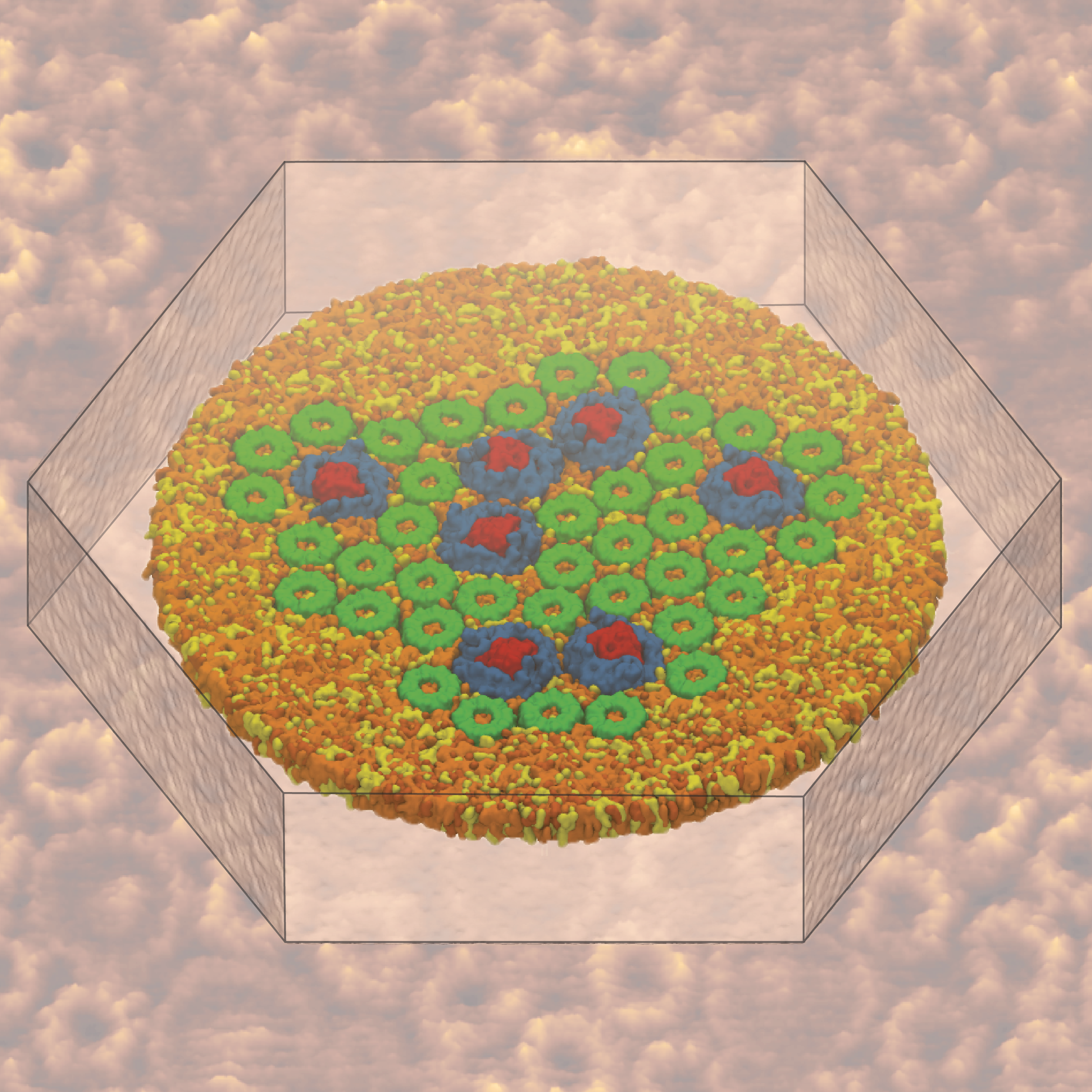
Nanoengineers building nanodevices achieve technological solutions at scales of 100 nanometers or 0.0001 mm. Nanoengineering is a brand new human technology, just a few decades old. In living cells, nanoengineering solutions are actually a few billion years old and therefore much more intricate. An impressive example is the nuclear pore, hundreds to thousands of which dot the nuclear membrane that separates in eukaryotic cells the genome and its molecular control factors from the cytoplasm of the cell. Only since very recently could cell biologists begin to resolve the molecular architecture of the nuclear pore. Given the pore's many-fold functions, like letting small molecules pass easily, but larger ones only as cargoes of special proteins, the transport factors, or adapting the pore size when large cargoes need to pass, the architecture of the nuclear pore is complex, involving an assembly of hundreds of proteins. The interior of the pore is filled with 600 amino acid-long "finger" proteins tethered at the periphery. The finger proteins are largely disordered such that experimental methods lack resolving power and computational modeling is needed to figure out their dynamic arrangement and traffic control function, but such modeling was largely unfeasible; only a small fraction of the nuclear pore volume could be covered computationally. The advent of petascale computing increased the size-scale of biomolecular simulations hundred-fold and a recent report employing the programs NAMD and VMD took advantage of the new generation of computers, simulating the dynamic, disordered arrangement of nuclear pore proteins. The simulations, still at an early stage, suggest a detailed, atomic level picture of the nuclear pore interior together with an explanation of molecular traffic control. More on our nuclear pore website.
image size:
199.7KB
made with VMD
Our body uses several defense mechanisms against seasonal flu, the common affliction caused by influenza viruses. By taking a yearly flu shot, our body's defense based on antibodies is trained and envoked. A defense system not based on antibodies acts at the very front line of influenza virus attack, namely the lungs. For this protection the body uses so-called lung surfactant proteins that coat the inner lining of the lungs to keep a wet film on the lung surface needed for oxygen-carbon dioxide exchange. The lung surfactant proteins also serve as police against influenza viruses. For this purpose the lung surfactant protein D (SP-D) recognizes a protein component of the virus surface, namely hemagglutinin, and handcuffs the sugar molecules bound to hemagglutinin. A previous experimental-simulation study (see October 2012 highlight) found that SP-D of pigs exhibits a stronger inhibitory activity against influenza A virus in this regard than does human SP-D. In a recent study, researchers have now boosted the protective ability of human SP-D by introducing mutations. Molecular dynamics simulations using NAMD suggest that the mutated human SP-D employs a different and stronger blocking mechanism on the active site of influenza A virus than native SP-D does. Combined with experimental results, the simulations suggest a mechanism through which SP-D acts, namely, by handcuffing viruses together and, thereby, preventing viral entry into cells. The findings from this research might lead to a new protection against seasonal flu, namely a nasal spray containing mutated lung surfactant proteins that strengthen a person's armada of defense proteins on the lung surface. More on our lung surfactant protein website.
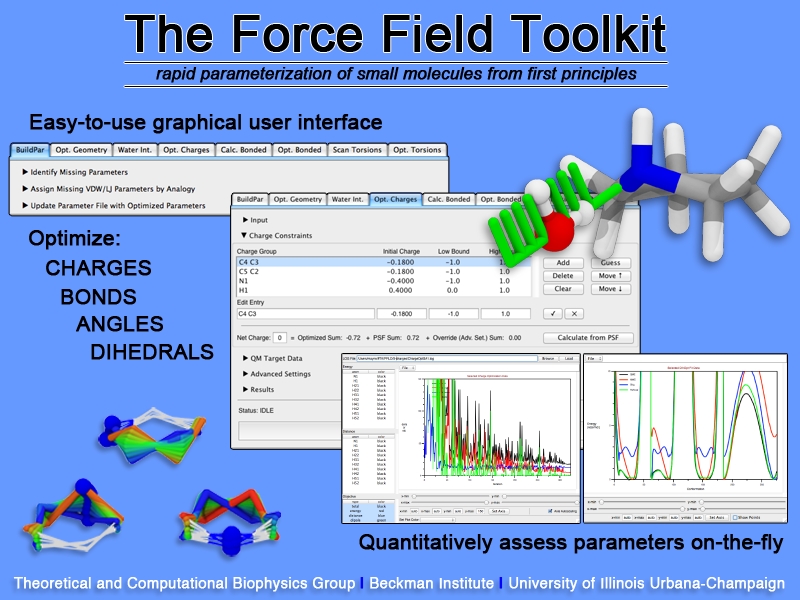
image size:
307.6KB
Structural biologists are increasingly turning to simulation methods to investigate the connections between molecular structure and biological function. Classical molecular dynamics (MD) simulations, such as those performed by the simulation software NAMD, rely on potential energy functions requiring parameters to describe atomic interactions within the molecular system. While these parameters are available for the most commonly simulated biopolymers (e.g., proteins, nucleic acids, carbohydrates), many small molecules and other chemical species lack adequate descriptions. The complexity of developing these parameters severly restricts the application of MD technologies across many fields, including most notably drug discovery. Recently, researchers have developed software, the Force Field Toolkit (ffTK), that greatly reduces these limitations by facilitating the development of parameters directly from first principles. ffTK, distributed as a plugin for the molecular modeling softare VMD, addresses both theoretical and practical aspects of parameterization by automating tedious and error-prone steps, performing multidimensional optimizations, and providing quantitative assessment of parameter performance--all from within an easy-to-use graphical user interface. Additional information on ffTK, including documentation and screencast tutorials, can be found here.
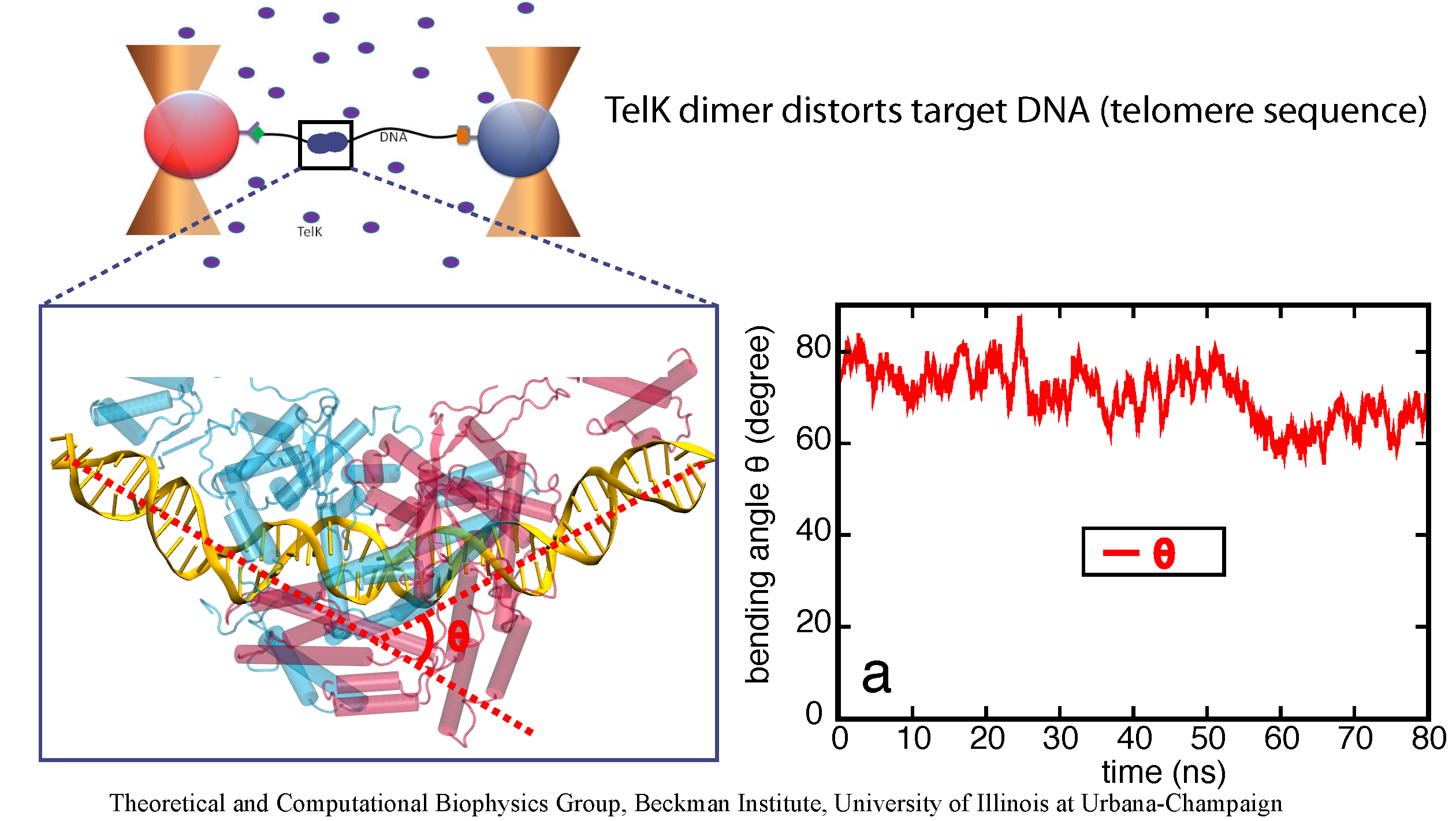
image size:
651.0KB
made with VMD
DNA, a long linear molecule, is the carrier of genetic information. In the cell, each DNA molecule is packaged in a structure called chromosome. The ends of linear chromosomes are capped by structures known as telomeres to prevent fusion with neighboring chromosomes. Telomeres are maintained by an enzyme called telomerase during DNA replication. In order to do so, telomerase has to find the telomere region on DNA quickly and precisely. One telomerase is the protelomerase TelK, which binds to the ends of DNA, cleaves DNA strands and refolds cleaved DNA ends into hairpin telomeres in linear chromosomes of prokaryotes and viruses. Previous studies have shown that TelK is only active as a dimer. In a recent study, researchers investigated the target-search mechanism of protelomerase TelK through single-molecule experiments and molecular dynamics simulation. It was revealed that as a monomer, TelK undergoes one-dimensional diffusion along non-specific DNA (without telomere sequence), and is able to bind to the target site preferentially. There, the target-immobilized monomer waits for a second binding partner to form an active protein complex. More on our TelK website.
Human immunodeficiency virus type 1 (HIV-1) is the major cause of AIDS, for which treatments need to be developed continuously as the virus becomes quickly resistant to new drugs. When the virus infects a human cell it releases into the cell its capsid, a closed, stable container protecting the viral genetic material. However, interaction with the cell triggers at some point an instability of the capsid, leading to a well timed release of the genetic material that merges then with the cell's genes and begins to control the cell. The dual role of the capsid, to be functionally both stable and unstable, makes it in principle an ideal target for antiviral drugs and, in fact, treatments of other viral infections successfully target the respective capsids. The size of the HIV-1 capsid (about 1,300 proteins), and its irregular shape had prevented so far the resolution of a full capsid atomic-level structure. However, in a tour de force effort, groups of experimental and computational scientists have now resolved the capsid's chemical structure (deposited to the protein data bank under the accession codes 3J3Q and 3J3Y). As reported recently (see also journal cover), the researchers combined NMR structure analysis, electron microscopy and data-guided molecular dynamics simulations utilizing VMD to prepare and analyze simulations performed using NAMD on one of the most powerful computers worldwide, Blue Waters, to obtain and characterize the HIV-1 capsid. The discovery can guide now the design of novel drugs for enhanced antiviral therapy. More information is available on our virus website, in video, and in a press release.

Rabbit hemorrhagic disease is extremely contagious and associated with liver necrosis, hemorrhaging, and high mortality in adult rabbits. First described in China in 1984, within a few years, rabbit hemorrhagic disease spread to large parts of the world and today threatens the rabbit industry and related ecology. The disease is caused by a virus, aptly named rabbit hemorrhagic disease virus. As reported recently, a group of experimental and computational researchers combining crystallography, electron microscopy and data-guided molecular dynamics simulations utilizing NAMD determined an atomic model of the capsid, namely the protein shell that surrounds the genetic material of the virus. The capsid simulations involved 10 million atoms and have become feasible only through Blue Waters, a brand new petascale supercomputer. The atomic model, analyzed by means of VMD, recently adapted to studies of very large structures, resolves the structural framework that furnishes both mechanical protection to the viral genes as well as a quick release mechanism after a virus enters a host cell. Researchers can use the detailed knowledge of the capsid structure to develop vaccines against rabbit hemorrhagic disease. More information is available on our virus website and in a news story.
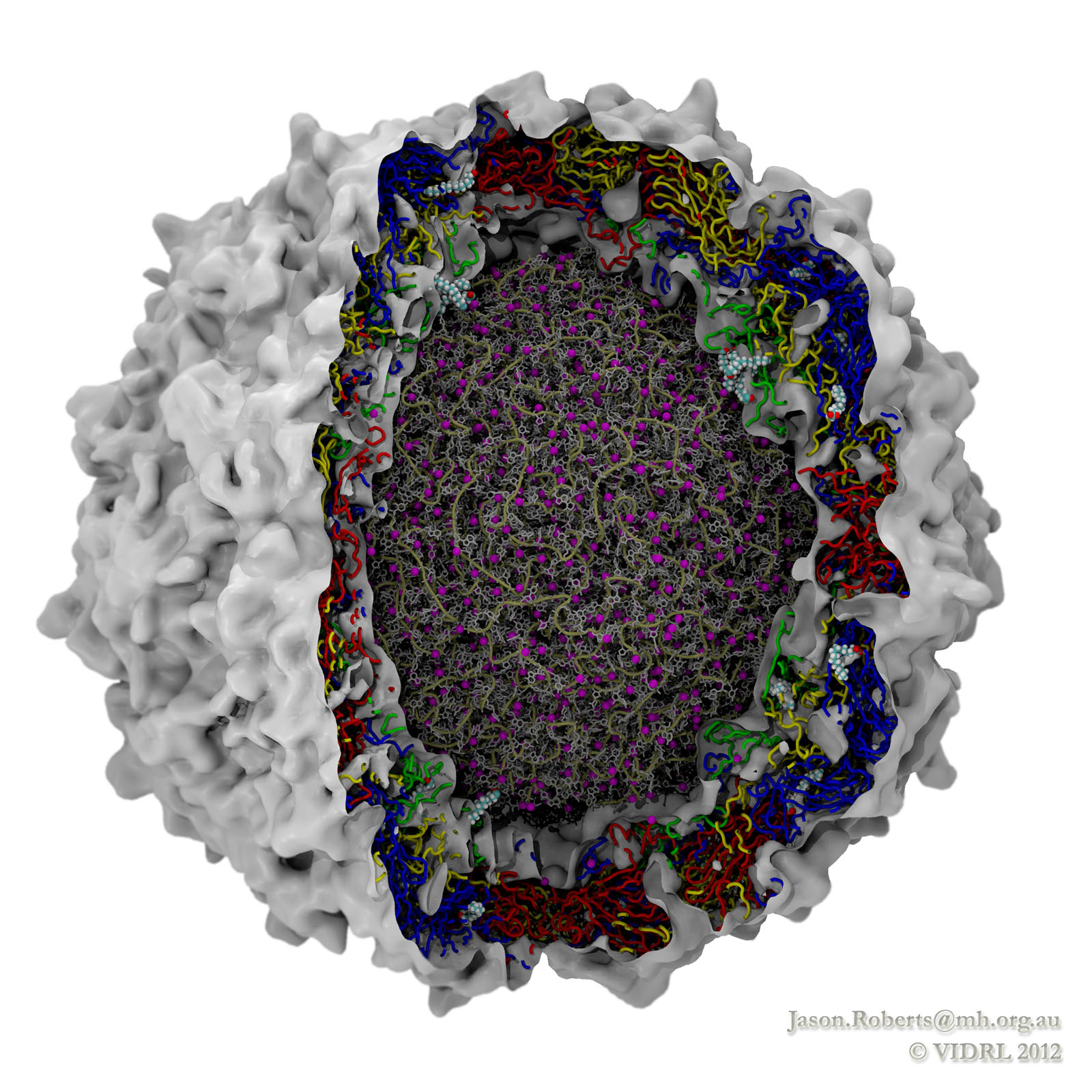
While a global campaign to eradicate polio has resulted in only 212 cases reported worldwide in 2012, there is a continuing risk of vaccine-resistant strains emerging. In addition, as one of the most studied viruses of all time, polio makes an excellent model for understanding similar diseases. Researchers at the Victorian Infectious Diseases Reference Laboratory have reconstructed and simulated a complete polio virus, including both the genomic payload RNA core and its surrounding protein capsid. The polio virus model is being used as a basis for understanding antiviral drugs, virus infection, and to learn how to model related viruses such as Enterovirus 71. The model was constructed in VMD and simulations of 3.7 million atoms were performed using NAMD on the BlueGene/Q supercomputer at the Victorian Life Sciences Computation Initiative. Rhombicdodecahedral periodic boundary conditions were used to reduce the volume of water needed to solvate the virus, and the symmetry of the virus capsid was exploited to study an antiviral drug binding at 60 identical locations simultaneously, avoiding artifacts that could have arisen from a truncated structure. See the researchers' poliovirus simulation website for a movie and additional information.
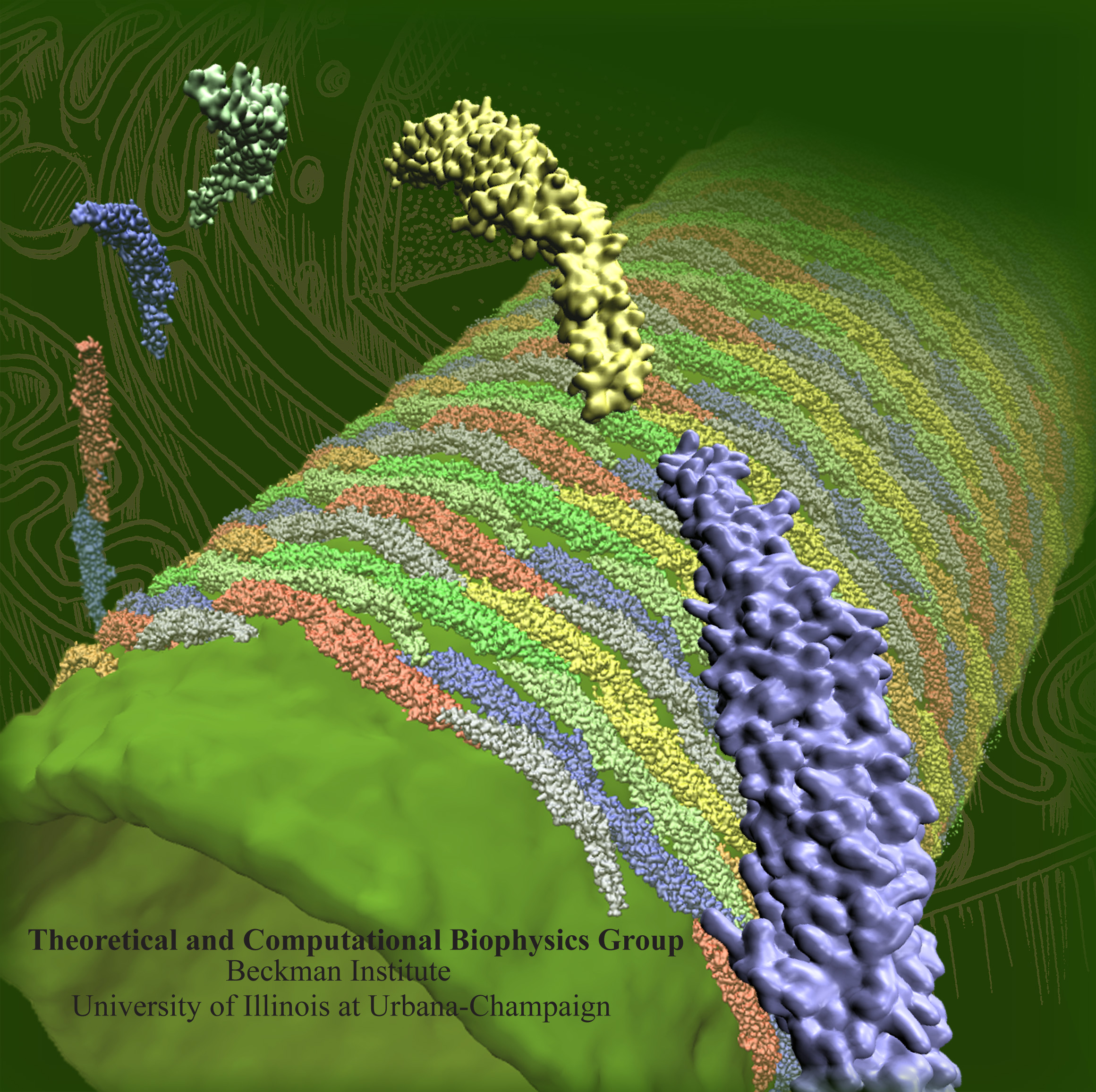
image size:
1.3MB
Olga Svinarski and VMD
The cells of higher life forms, so-called eukaryotic cells, are subdivided through many internal membranes made of lipid bilayers. The internal membranes assume numerous shapes, like spheres, tubes or parallel sheets. Outside of cells, biological membranes adopt usually flat shapes and the question arises, how do eukaryotic cells sculpt their inner membranes? The question of flat membrane sculpting is particularly interesting also as mature cells constantly produce new membrane shapes, for example spherical vesicles filled with certain biomolecules destined for release into the extracellular space, a process called exocytosis. The cell has many mechanisms available for sculpting its membranes, one of them relying on proteins called BAR domains that act from the surface of lipid bilayers. Molecular modeling with NAMD and VMD has provided valuable views of BAR domains at work in case of the so-called N-BAR family (see the earlier highlights Protein Teamwork, Jun 2009 and Proteins Sculpting Cell Interior, Sep 2008). Researchers report now an extension of the earlier studies to the F-BAR domain family of membrane sculpting proteins. The new modeling work is particularly exciting as it can be directly compared to electron microscopy images of membrane tubes sculpted from flat membranes in experiments done outside of cells. The new studies reveal how F-BAR domains sculpt tubular membranes through the shape of dimerized domains and through F-BAR domains not acting individually, but as an army of F-BAR domains adopting an ordered formation on one side of the membrane. More on our F-BAR domain web page.
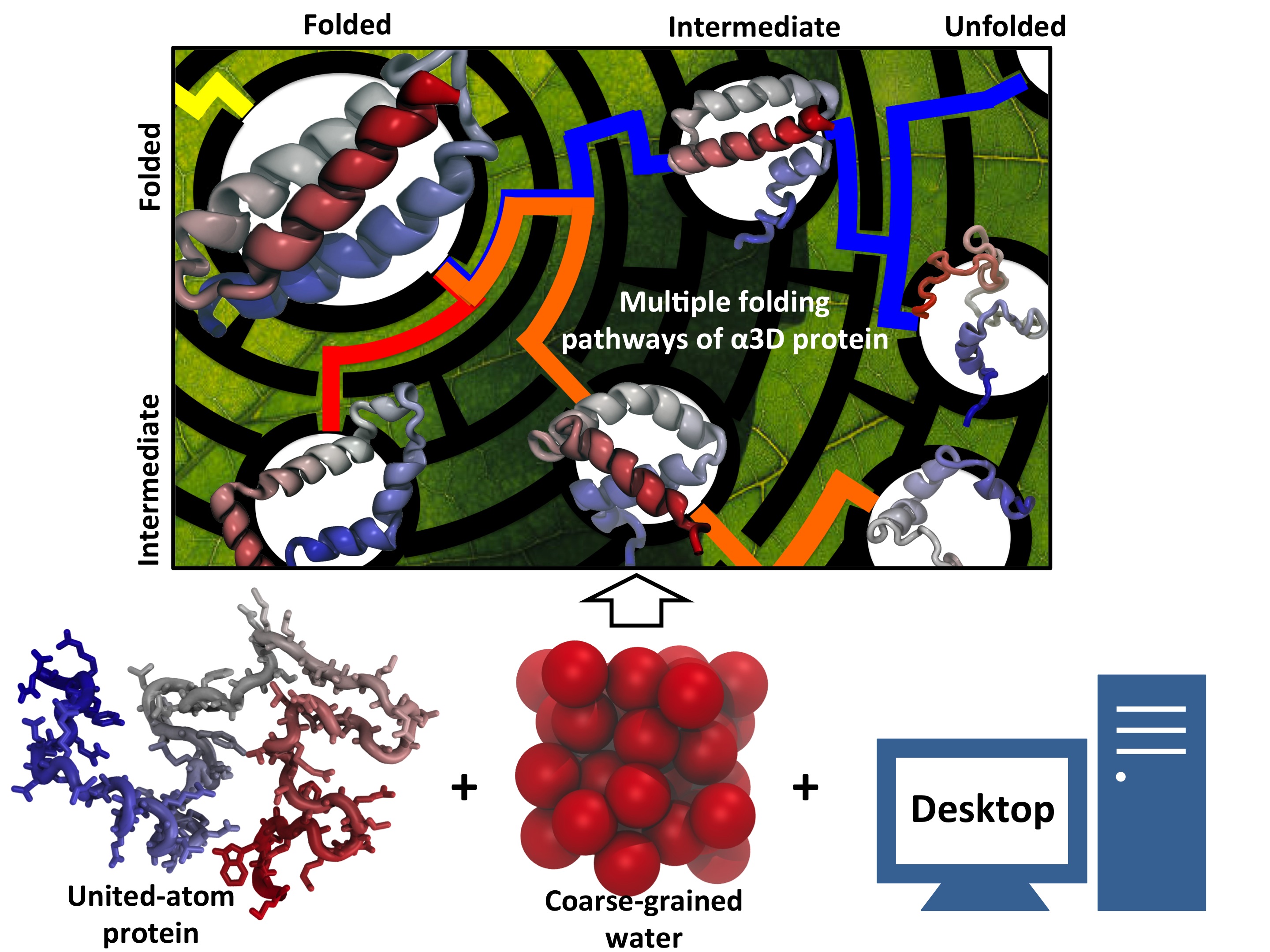
Every living cell relies on proteins to carry out its functional tasks; every protein needs to assume a proper shape in order to be operational for these tasks. How a protein, composed of a particular sequence of amino acids, could find its way to a proper shape is a fundamental, yet mysterious biological process. Researchers have sought to unravel atomistic details of protein folding processes through computer simulations, but modeling such processes is computationally demanding. It was only recently that some researchers have been able to observe in some case how proteins fold, but needed for the purpose the fastest computers available today. One of these computers is Anton, the expensive special-purpose supercomputer available essentially only to a single research group. Is there an affordable way to simulate protein folding? One solution could be coarse-grained methods. These methods save tremendous computational effort by replacing computational models that include all atomistic detail. However, the simplified models need to include a sufficiently accurate description of proteins for modeling folding processes. As reported recently, researchers have overcome the challenge by combining atomistic and coarse-grained descriptions. The new method is fast enough to follow movements of proteins long enough to see them fold, while requiring only readily available computer powers. The new method allowed researchers to analyze complete folding events for seven proteins, including a protein, called α3D (see movie, 11.5 M), that is one of the largest proteins ever folded computationally. More on our hybrid-resolution model website.

image size:
77.5KB
made with VMD
Did you get your flu shot this year? Influenza is a leading cause of preventable death in the industrialized world, representing hundreds of billions of dollars in healthcare expenditures and loss of economic production. While the yearly influenza vaccination is nearly 90% effective at limiting infections in populations less than 65 years of age, there is insufficient evidence regarding the effectiveness of the flu shot for the elderly population, whose immune systems may not mount an adequate antibody response to vaccination. Beyond vaccination, front-line therapies such as the neuraminidase inhibitors Tamiflu and Relenza have proven to be of limited effectiveness due to the evolution of drug-resistant influenza mutants. Therefore, a need exists for the development of new therapies to circumvent these resistance mechanisms. Computational biologists employing NAMD and VMD used molecular simulations to uncover the key role that water plays in mediating how well antiviral drugs can bind to proteins of the influenza virus. This investigation, reported recently, reveals that amino-acid mutations responsible for drug resistance act by reshaping the local electric field and also by permitting infiltration of water within the otherwise hydrophobic drug binding pocket. These mutations thus induce drug resistance in much the same way as inverting the polarity of a magnet can repel rather than attract. These findings are expected to help guide the design of novel drugs with increased antiviral efficacy. Additional details about this study can be found here.
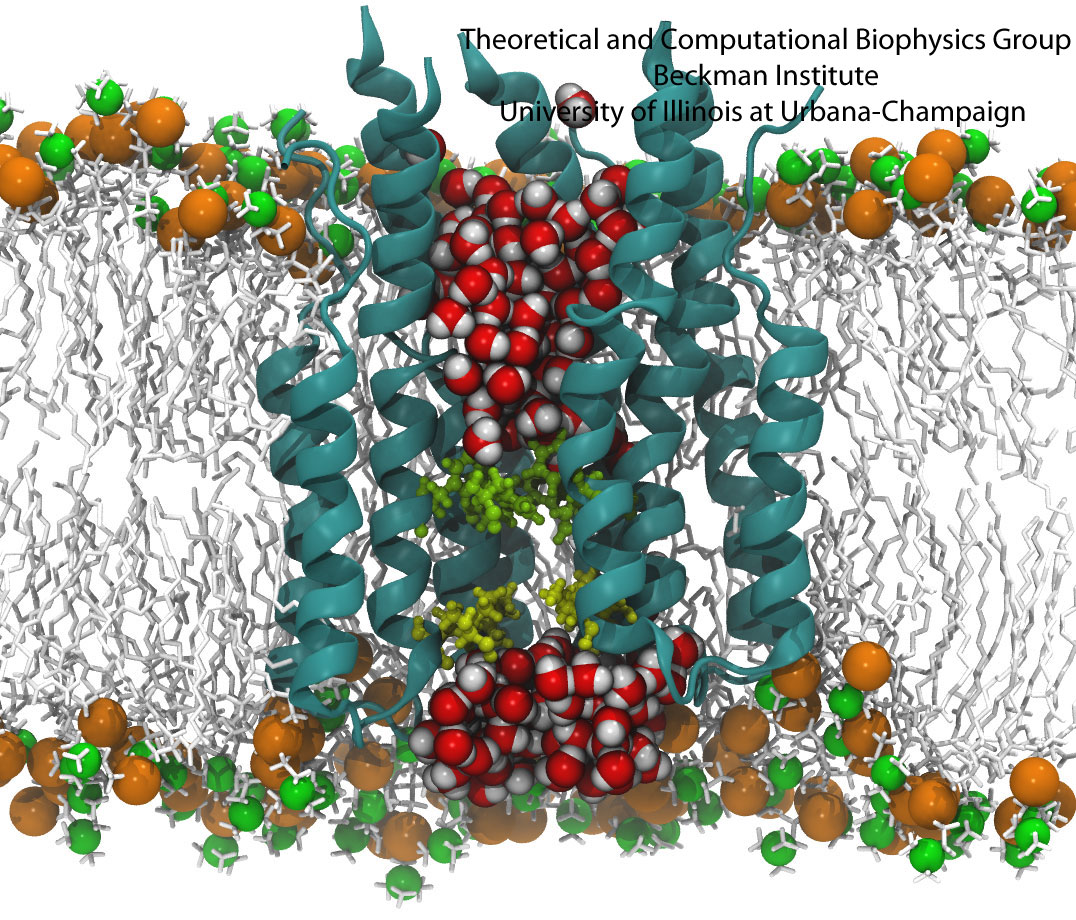
All living systems contain proteins whose job is to move ions across a lipid membrane. Even viruses encode ion transport proteins, which they need to complete their lifecycle and release themselves from infected cells. Such proteins, called viroporins, usually consist of small subunits of one or two helices that can self-assemble in a lipid bilayer into a pore-like structure. Although in some cases, the resulting structures resemble the well-ordered, selective ion channels in higher organisms, often they take on a more disordered character, forming pores with variable numbers of subunits, which adapt their structure and behavior to the environment in which they find themselves. This inherent flexibility and disorder makes it very difficult to produce high-resolution crystal structures of viroporins, which is unfortunate, since they could offer attractive drug targets for new antiviral therapies. Computational modelling and molecular dynamics simulations can help fill in the gaps in our structural knowledge of viroporins, and provide plausible 3-D models for visualization and drug design. In a recent publication, scientists published models of the p7 viroporin found in Hepatitis C virus. MD simulations of these models revealed that p7 can form stable pores with 4 to 7 subunits, with a bias towards 6 or 7 subunits, and that the p7 oligomers are highly flexible in adapting to different membrane thicknesses. These simulations also suggested that specific amino acids in certain places in the structure could play a role in controlling the ion permeability of p7. More details can be found on our p7 website.
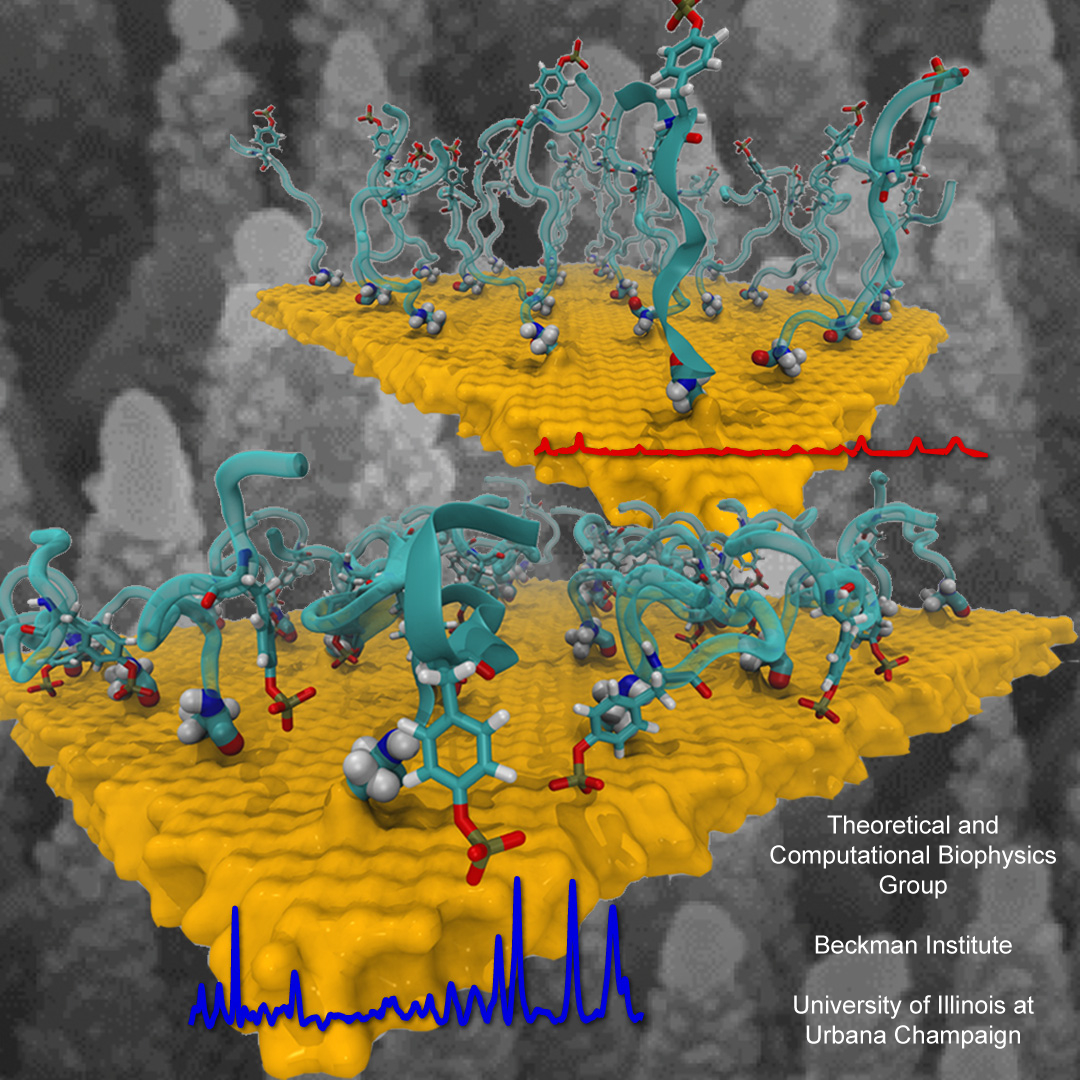
Nanoengineering permits the manufacturing of sensors of unprecedented accuracy to detect biomolecules at very low concentrations as they arise, for example, as signals in living cells. In an important type of cellular signaling, proteins are modified through addition of a phosphate group by other proteins, so-called kinases. Kinases are involved in various types of cancers; therefore, bioengineers seek to develop a nanosensor to detect kinase activity. As described in a recent report, they grafted short peptides, containing a tyrosine amino acid, on nanometer scale gold surfaces. Phosphate groups are negatively charged, and as the groups are transferred from the kinases to the peptides tyrosine, the overall charge of the grafted peptides increase. Bioengineers detected then the phosphorylated peptides by applying electrical fields that would drive the charged phosphate group towards the surface or away from it, depending on the voltage polarity; the resulting conformational change of peptides can be recognized by shining light on the nanosensors as optical properties of molecules near metal surfaces are amplified. In order to make the nanodevice really work, the bioengineers needed to optimize the peptide sequence, know how phosphorylation and voltages alter the near-surface conformation of the peptides and how to interpret the measured optical signals. In other words, they needed a microscopic view of the nanodevice! Such view was achieved through molecular dynamics simulations using NAMD and VMD following in the footsteps of similar earlier uses of such simulations as a computational microscope (see Diet and DNA, Sep 2011; Bumpy DNA, Feb 2009; Stretchable DNA, Nov 2005). The combination of nanoengineering and molecular dynamics simulations produced indeed a satisfactory kinase sensor prototype. For more information, visit our kinase sensor website.
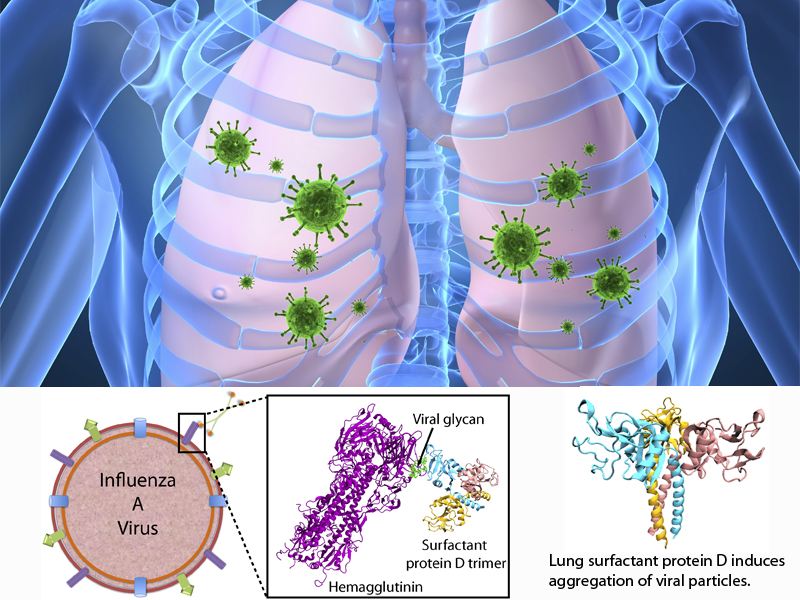
image size:
76.8KB
made with VMD
The fall flu season is coming. It is time to get your flu shot! Many people may still remember the influenza A H1N1 flu ("swine flu") pandemic of 2009, which caused 280,000 deaths worldwide. The best way to prevent the flu is to get vaccinated with a flu shot or use the flu nasal spray vaccine. However, rapid evolution of the flu virus constantly requires new vaccines. Fortunately, the immune system has several defensive mechanisms in the lung to clear inhaled pathogens. One of these mechanisms involves surfactant proteins which induce aggregation of viral particles and, thereby, prevent infection, serving as a front-line host defense. Recently, researchers found that surfactant protein D (SP-D) from pigs exhibits particularly strong inhibitory activity, more so that human SP-D. This discovery leads researchers to investigate SP-D structure-related antiviral activity. In a recent experimental-simulation study, crystallographic analysis of pig and human SP-D showed that a loop involved in viral binding on pig SP-D is longer than the respective loop on human SP-D; molecular dynamics simulation revealed that the longer loop of pig SP-D has higher flexibility than that of human SP-D, suggesting that the flexible loop region could facilitate strong binding of SP-D to virus. Based on this finding, one can develop new nasal spray anti-flu protection through other structural modification of human lung surfactant proteins. More on our lung surfactant protein website.
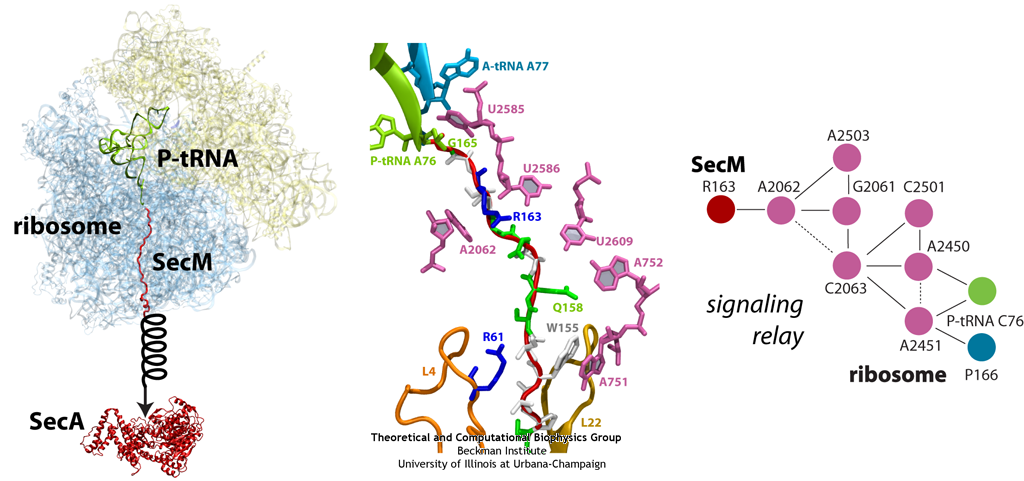
image size:
299.9KB
made with VMD
The ribosome functions as a cellular protein factory, synthesizing practically all the proteins in the cell based on blueprints read from DNA (see the April 2012 highlight and Dec. 2009 highlight). However, unlike an assembly line, the ribosome has no foreman directing it. Instead, regulation of protein synthesis is managed by a number of external, and internal, signals. For example, the protein TnaC halts its own synthesis in the ribosome to promote that of another protein (see the May 2010 highlight). Similarly, synthesis of the protein SecA, a translocase that aids in pushing newly made proteins across membranes, is controlled through the nascent protein SecM. Regulation of SecA levels is the only function of SecM, which is degraded as soon as it leaves the ribosome. It is the stalling of one ribosome by SecM that provides enough time for secA, which resides on the same messenger RNA as secM, to be translated by a second ribosome, thus upregulating SecA production. When enough SecA has been produced, it pulls on the portion of SecM outside the ribosome, relieving its stalled state. The critical interactions that cause stalling have now been identified through a combination of molecular dynamics and cryo-electron microscopy via MDFF and NAMD. As recently reported, these interactions form a relay connecting SecM in the exit tunnel to the ribosome's key catalytic center, preventing synthesis and thus explaining how SecM stalls inside "its" ribosome. Additionally, steered MD simulations revealed how SecA can cause the nascent SecM to become unstuck, by breaking those same interactions. More details are provided on our ribosome website.
Proteins are the biological workhorses in living cells. For example, they respond to external signals arriving at the cell surface or transport cargo, much larger than themselves, from one place to another in the cell. However, before a protein can carry out his job, it must first assume the proper shape. Proteins are long polymers of twenty different amino acids linked in a linear sequence; the latter is particular for each protein. It is still a mystery how a protein folds into the proper shape based on its sequence. Scientists hope that one day they can "watch" this folding process for any given protein. The dream has been realized, at least partially, through the use of computer simulation. After tackling the protein-folding problem already computationally for two small proteins (see May 2008 highlight and Nov 2009 highlight), researchers have now successfully visualized the complete folding process of a relatively large protein, the so-called λ-repessor (see movie, 7.8 MB). In fact, it is one of the largest proteins folded to date using a computer. As reported recently, simulations carried out with the program NAMD, as well as simulations carried out on a special purpose supercomputer, Anton, achieved to follow λ-repessor's folding movement for more than 0.0001 seconds, long enough to observe the protein assume its proper shape. More information is available on our protein folding website.
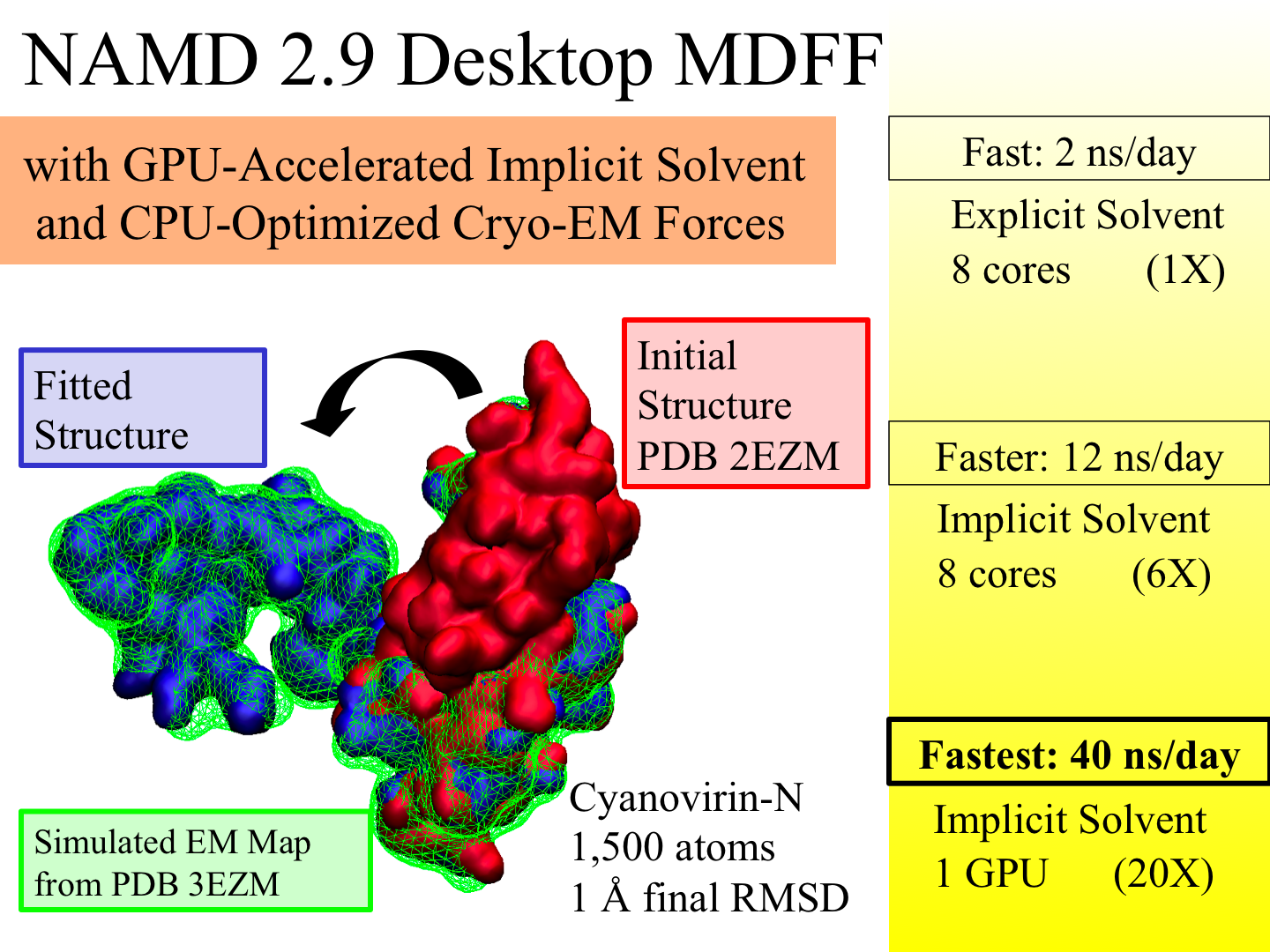
image size:
883.7KB
made with VMD
X-ray crystallography resolves the structures of the molecular machines in living cells at an atomic level of detail, but only in states that can be captured as crystals, which are often not functional states. Cryo-electron microscopy enables a more complete view of biomolecular conformational variability, but at lower resolution. The molecular dynamics flexible fitting (MDFF) method combines the atomic detail of crystallographic structures with lower-resolution cryo-electron microscopy to synthesize all-atom models of complex macromolecular aggregates such as the ribosome in multiple functional states. The 2.9 release of NAMD combines GPU acceleration of implicit solvent simulation, optimizations exploiting shared memory within a single machine, and a faster "lite" grid forces implementation to bring MDFF capability from the supercomputer to the desktop. VMD can connect to a running simulation to visually monitor the progress of the simulation or to interactively steer a molecule with either the mouse or a haptic (force-feedback) interface device. The convergence of methodology, software, and hardware advances thus opens what was once the domain of extremely expensive equipment to commodity computers.

made with VMD
Viruses reproduce by splicing their genetic material into a host cell, causing the cell to manufacture new viruses. This genetic material is protected outside of the host cell by a protein capsid, which disassembles inside a new cell to complete the infection process. Simulation of viral infection has progressed significantly since the first all-atom virus simulation was done with NAMD in 2006 and is one of the driving biomedical projects for the software. A new collaboration with the Pittsburgh Center for HIV Protein Interactions has applied molecular dynamics flexible fitting to construct the first all-atom structure of an HIV virus capsid in its tubular form (shown). This structure is now being simulated as one of six early science projects on the Blue Waters petascale supercomputer being installed at Illinois. These large-scale simulations are enabled by the 2.9 release of NAMD, which includes a new high-performance interface to the Cray Gemini network of Blue Waters. Smaller simulations may also leverage petascale computing through a new replica-exchange framework that supports parallel tempering and integrates with the collective variables module for umbrella sampling conformational free energy calculations. GPU acceleration enhancements include minimization and implicit solvent support as well as exploitation of shared memory, extending performance gains to the desktop.
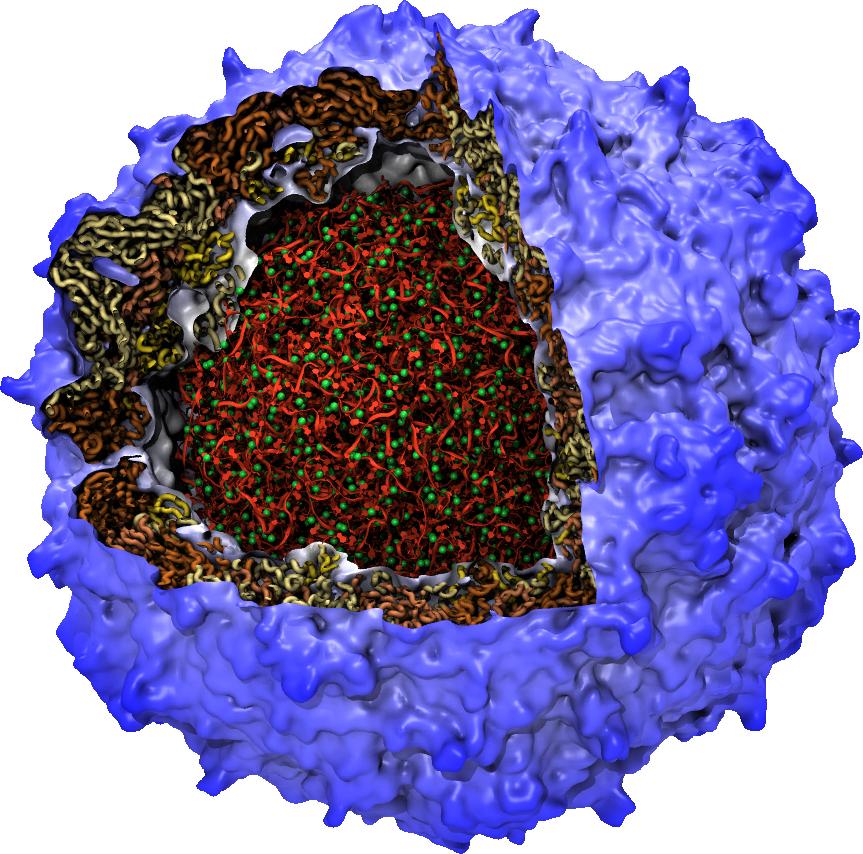
The surfaces of biomolecules are alive with activity, with surface shape and electrostatic interactions leading them to interact with each other. The recent VMD 1.9.1 release includes a new "QuickSurf" graphical representation for molecular surfaces that allows the dynamics of large biomolecules to be animated interactively for the first time. VMD 1.9.1 even enables surface representations for many-million atom complexes. QuickSurf uses fast algorithms, GPU computing techniques, and multi-core CPUs to achieve astonishing performance. The algorithms behind QuickSurf have been recently reported. VMD 1.9.1 adds many other new features including a new Force Field Toolkit (FFTK) plugin that assists researchers in development of CHARMM force field parameters, a new NetworkView plugin for mapping and displaying networks on 3-D molecular structures, an updated ViewChangeRender plugin for making sophisticated demonstrations and movies, and a new VMD remote control tool that allows VMD sessions to be controlled from wireless touch sensitive phones and tablet devices. For more on these and other new features of VMD see the VMD 1.9.1 release page.
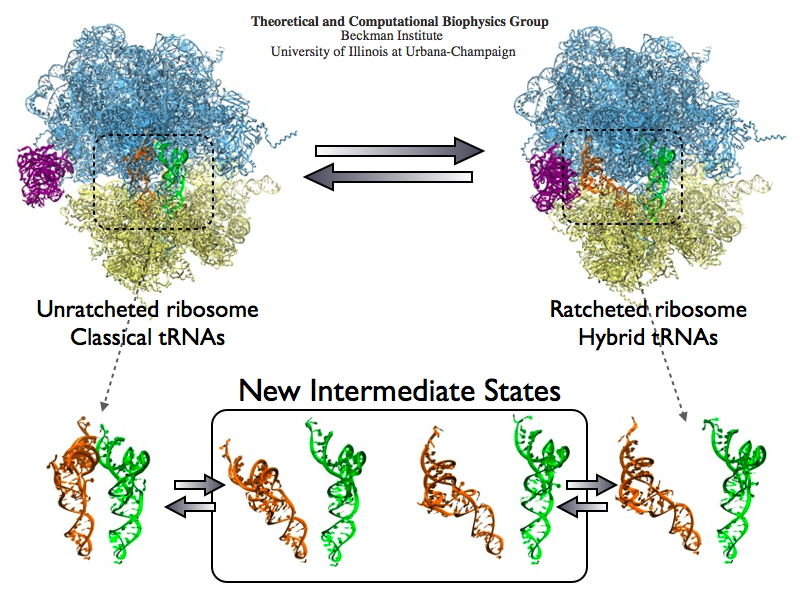
image size:
488.4KB
made with VMD
The ribosome is the protein assembly line in all living cells. The building material for new proteins is supplied by RNA molecules, called tRNA. They enter and move through the ribosome, each adding a new amino acid to the nascent proteins according to the genetic sequence provided through so-called messenger RNA. During the tRNAs' translation through the ribosome, the ribosome itself is not static either. A ratcheting motion and other large scale motions can be observed. However, the exact tRNA and ribosome motion were not clear. Using images from cryo-electron microscopy, MDFF, a computational method based on NAMD, allows one to see the moving parts within the ribosome in great detail. MDFF (see the June 2008 highlight) already provided crucial and unique insights into different aspects of protein synthesis, such as translational arrest of the ribosome by a nascent chain or translocation of an emergent protein across a membrane. In the work reported recently, MDFF revealed the presence of previously unseen intermediate states of the ribosome and its bound tRNAs during the ratcheting motion. A thorough analysis of these states pictures the ribosome as a molecular machine using Brownian motion for its function. More on our ribosome website.
image size:
419.0KB
made with VMD
Voltage-gated ion channels, present in the membrane of excitable cells, control the ionic concentrations of the cellular environment by maintaining a potential difference of -100 mV between inside and outside of the cell membrane. Voltage-sensing occurs through distinct protein modules, known as voltage-sensor domains, four of which surround the main conduction pathway in potassium channels. Mutation of a certain amino acid on the voltage sensor domain turns these protein modules into cation channels, known as omega pores, which allow conduction of ions only when the main pathway is closed. Omega pores closely resemble the long-sought voltage-gated proton channels, which were recently identified to follow the same voltage-sensing mechanism as voltage-gated cation channels. In a recent report, researchers have visualized the twisted permeation pathway of the ions through omega pores using the molecular dynamics program NAMD. The simulations revealed a narrow constriction region lined by negatively charged amino acids, acting as a selectivity filter that prefers passage of positively charged ions through the pore. For more detail, see our potassium channel website .
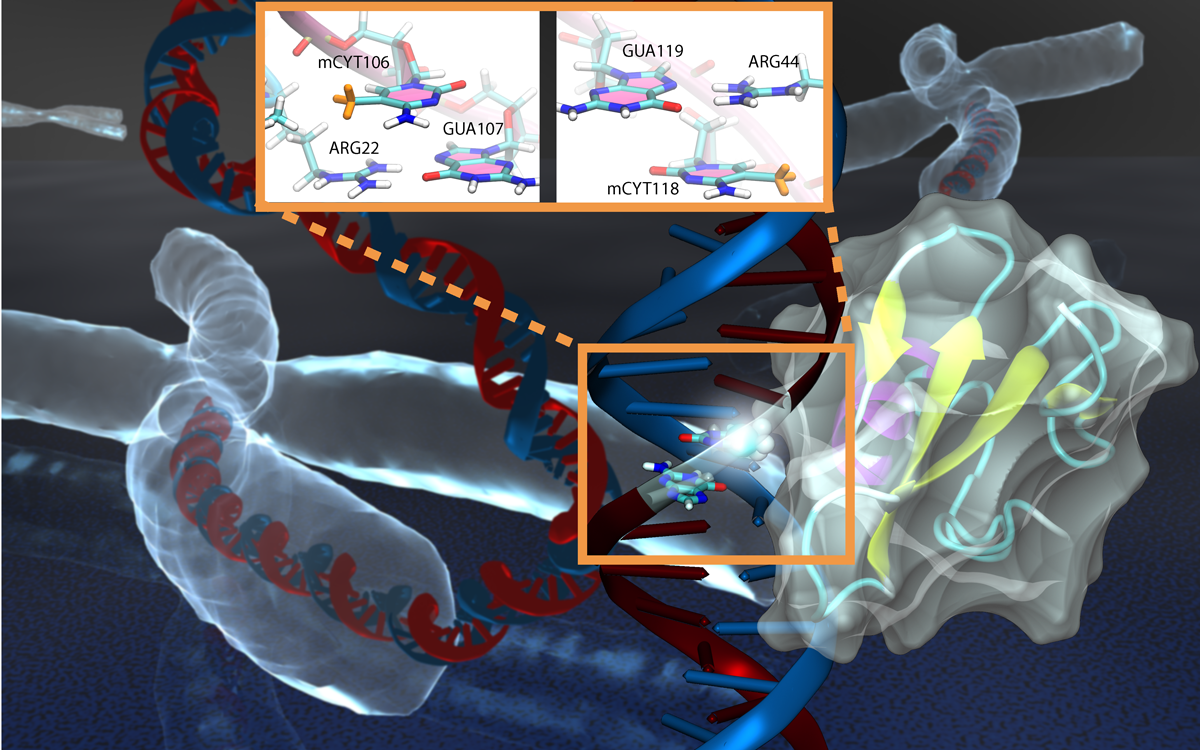
image size:
85.6KB
made with VMD
Genes encoded in DNA sequence give a complete set of instructions for the development of a new organism. However, an organism, like the human body, develops also over a life time adapting to environment and experience, for example to diet and exercise. Recently, researchers found that such factors act through so-called epigenetic mechanisms that alter an organism's development without altering DNA sequence. One such mechanism involves DNA methylation, a chemical modification of one of the four bases of DNA, cytosine, that replaces a hydrogen atom with a methyl group. There are several ways that DNA methylation exerts its biological function, bringing about a long-time adaptation of an organism to its environment, in some cases even across generations. Our previous experimental and computational studies (see Sep 2011 and Feb 2009 highlights) indicated that methylation changes mechanical properties of DNA which can affect gene expression. DNA methylation can also inhibit gene expression by impeding proteins that control the translation of DNA sequence into protein synthesis. One mechanism involves DNA methylation sites recruiting genetic control proteins that inhibit DNA expression through their local presence. In a recent study, computational biologists performed MD simulations with NAMD along with quantum chemistry calculations to determine recognition of methylated DNA by proteins. The simulations revealed how a certain genetic control protein, called methyl-CpG binding domain protein, acts in tandem with methylated DNA like a key and a lock, methylated DNA and protein perfectly matching each other. More details can be found on our methylated DNA website.
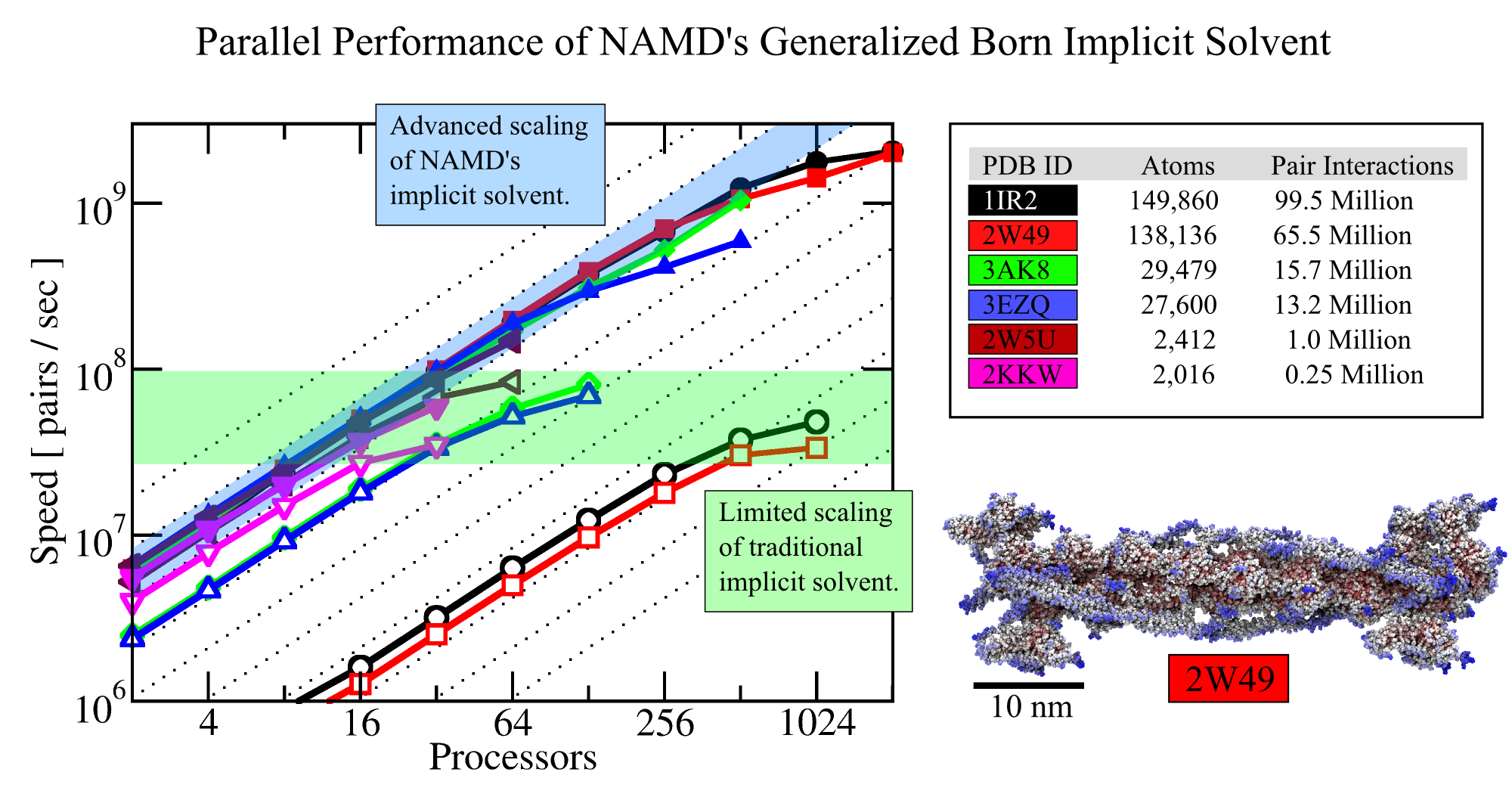
image size:
573.0KB
Olga Svinarski and VMD
Water is ubiquitous; living cells and the cells' biomolecules are greatly influenced by water. Accounting for water in computational modeling, a key access route to imaging cell processes, comes at a stiff price: up to 90% of the simulation volume needs to be a water bath embedding simulated biomolecular machinery. The sheer number of water molecules arising here, sometimes millions, and the strong hydrodynamic drag resisting biomolecular dynamics, slows down simulations tremendously. Computational biologists had invented a means to replace water by a kind of continuous ether, so-called implicit solvent, that accounts for key electrostatic features of water around biomolecules, but does not slow down simulations. Computational hinderance through explicit water is getting worse with increasing size of biomolecular machines, but so far the implicit solvent description could not be effectively employed to model large systems using supercomputers. In a recent study, the situation has been repaired and the use of an implicit solvent in molecular dynamics simulations of systems as large as the whole ribosome (containing 300,000 ribosome atoms and eliminating 2,700,000 water atoms) have been documented. In a further study, the implicit solvent description has been extended to account also for hydrophobic interactions at biomolecular surfaces; the extension took advantage of delegating calculations both to GPU and CPU as the latter is more suitable for the evaluation of surface interactions. More information regarding NAMD's parallel computer-ready implicit solvent can be obtained from the NAMD website.
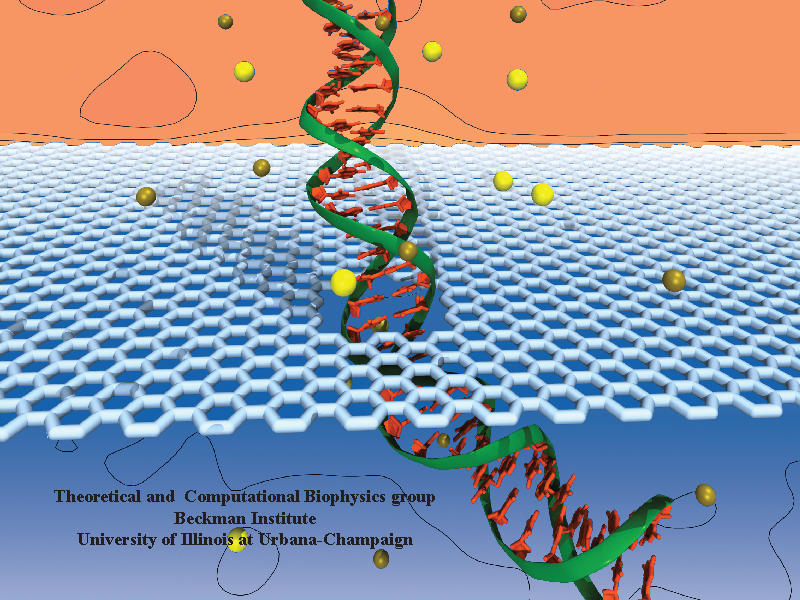
Threading DNA electrically through nanometer-sized pores, so-called nanopores, holds promise for detecting and sequencing DNA (see Nov 2005 and Oct 2004 highlights). Nanopore measurements tend to be the more sensitive the smaller the pores are. The material graphene, which is just one atom thick and looks like a two-dimensional ``honeycomb" made up of carbon atoms, offers the ultimate physical resolution for measuring DNA (the stacking distance between base-pairs in DNA is about 0.35 nm). As reported recently, molecular dynamics simulations using NAMD revealed the motion of DNA being threaded through graphene nanopores at atomic level resolution. Simulations not only agree qualitatively with previous experiments on DNA translocation through graphene nanopores, but go one step further than the experiments and suggest how individual base pairs can be discriminated. The recent computational study is one further example for the guidance that molecular dynamics simulations provide in nanosensor development (see a recent review). More information can be found on our graphene nanopore website.

Computer simulations of the biomolecular processes in human cells guide better understanding of health and disease as well as development of dietary supplements and pharmacological treatments. Such simulations are extremely demanding and, in fact, all too often still limited by technological feasibility. However, technological advances are being brought to bear on computer simulations in biomedicine through highly dedicated biomedical engineers, who have often speerheaded uses of new computer technologies such that they became available in biomedicine much sooner than in other fields. A case in point is solid state disk (SSD) technology that can serve as extremely fast and large computer memory. Conventional RAM (random access memory) is fast, but limited in size due to cost; the well known hard disks (HDs) can hold large data sets at an affordable price, but are slow. The new SSDs are in the middle ground, faster than HDs, slower than RAM, offering large data storage at an affordable price. Modern uses of SSDs in smart phones and tablets attest to the usefulness of SSDs. The biomolecular visualization and analysis software VMD in its next release (VMD 1.9.1) makes the power of SSDs as a huge, yet fast storage medium available to biomedical researchers. As reported, this will allow them to view and analyze through VMD on the fly Gigabytes-to-Terabytes of simulation data, that are being raked into a computer at the rate of up to 4 Gigabytes per second (one high definition video of a long movie per second!). For more on this and other revolutionary features of VMD 1.9.1 see our VMD web site.
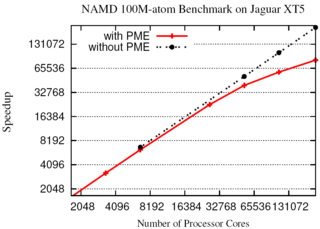
The NAMD molecular dynamics program excels at simulating, in atomic detail, the complex molecular machinery of living cells. NAMD now enables hundred-million-atom simulations using the full capabilities of the nation's fastest supercomputers due to parallel programming innovations reported in a paper at the SC11 supercomputing conference. While parallel supercomputers have massive amounts of memory in aggregate, any part of the machine can directly access only a small fraction. To fit the molecular blueprint of cellular structures such as the chromatophore in the memory of such machines, a compressed data format was developed that exploits the repetitive nature of proteins, nucleic acids, lipid membranes, and solvent. This format requires only a small amount of data to be copied to all parts of the supercomputer, while the remainder is read from disks in parallel and distributed across the machine. Simulation output and the balancing of work loads are similarly distributed. The Charm++ parallel programming system, on which NAMD is built, was also extended to allow the molecular structure and other data to be shared among the increasing number of cores in modern processors. A hundred-million-atom simulation can now run on supercomputers with only 2 gigabytes of memory per node, such as the IBM BlueGene/P. The enhancements are available to computational biologists world-wide in special memory-optimized versions of NAMD 2.8.
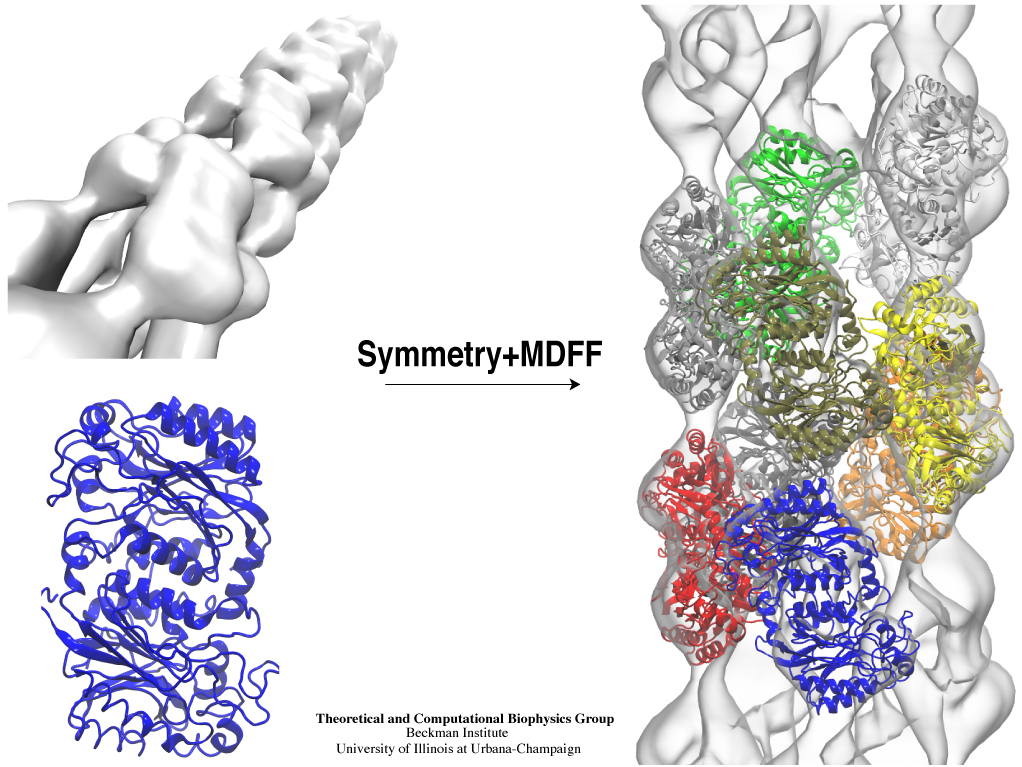
Inorganic nature brings about symmetry that one can admire, for example when one sees a polished diamond. Living nature, too, brings about symmetric structures; for example, many processes in living cells are carried out by highly symmetric protein complexes. In case of living cells symmetry comes about partially out of physical or geometrical necessity, but also partially out of its usefulness for the intended purpose. Hence, understanding symmetry in biology goes beyond related studies of the beauty of symmetry in the physical and mathematical sciences as one also asks: Does symmetry help? Put it another way: Symmetry in living cells is beautiful and useful! The proteins in symmetric complexes are often imaged by electron microscopy (EM), but unfortunately not at a resolution high enough to recognize chemical detail, as is needed in most cases, e.g., in case of the study of a symmetric multi-enzyme system. Computational biologists have developed a method to solve the resolution problem, using high resolution images obtained through X-ray scattering of related molecules and matching them through molecular dynamics using NAMD to the image seen in EM. This method, called molecular dynamics flexible fitting (MDFF) has been highlighted previously and is described on our MDFF website. As reported recently, MDFF has now been extended to determine the atomic-level structure of symmetric multi-protein systems. MDFF has been applied successfully to three highly symmetric multi-protein systems: (i) GroEL-GroES, a protein complex that assists proteins to fold properly into their native conformation; (ii) a nitrilase multi-enzyme system that converts massive amounts of molecules into forms more suitable for a bacterial cell; (iii) Mm-cpn, a protein complex supposedly involved in assisting protein folding in so-called archaebacteria. In all cases the symmetry of the protein complexes plays a key role. For more details, see the Method section of our MDFF website.

image size:
284.7KB
made with VMD
Bacteria contain the simplest photosynthetic machineries found in nature. Higher organisms like algae and plants practice photosynthesis in a more elaborate but principally similar manner as bacteria. But even for its simplicity, the bacterial photosynthetic unit is not without its unsolved mysteries. Take, for example, the crucial photosynthetic core complex, which performs light absorption and the initial processing of the light energy. In certain bacterial species, the core complex are each ring-shaped singlets, but in some other bacterias they can be 8-shaped doublets, formed by the association of two ring-shaped core complexes. The question arises: What holds doublets together. It turned out that doublets arise when bacteria bacterial contain two copies, i.e., twins, of an additional small protein called PufX. Recently, computational biologists and biochemists have come together to study PufX from different bacteria. They found an interesting trend showing that bacterial species with PufX that associates with another PufX protein also contain core complexes that form 8-shaped doublets. On the other hand, bacteria whose PufX is incapable of associating with another PufX have only singlets of ring-shaped core complexes. These new results resonate with a proposed role of PufX as the protein that holds two core complexes together to form 8-shaped doublets. Bacteria with PufX that cannot perform this role therefore only have singlets of ring-shaped core complexes. The needed simulations were done with NAMD. More details can be found on our photosynthetic core complex website and in a previous highlight on PufX.
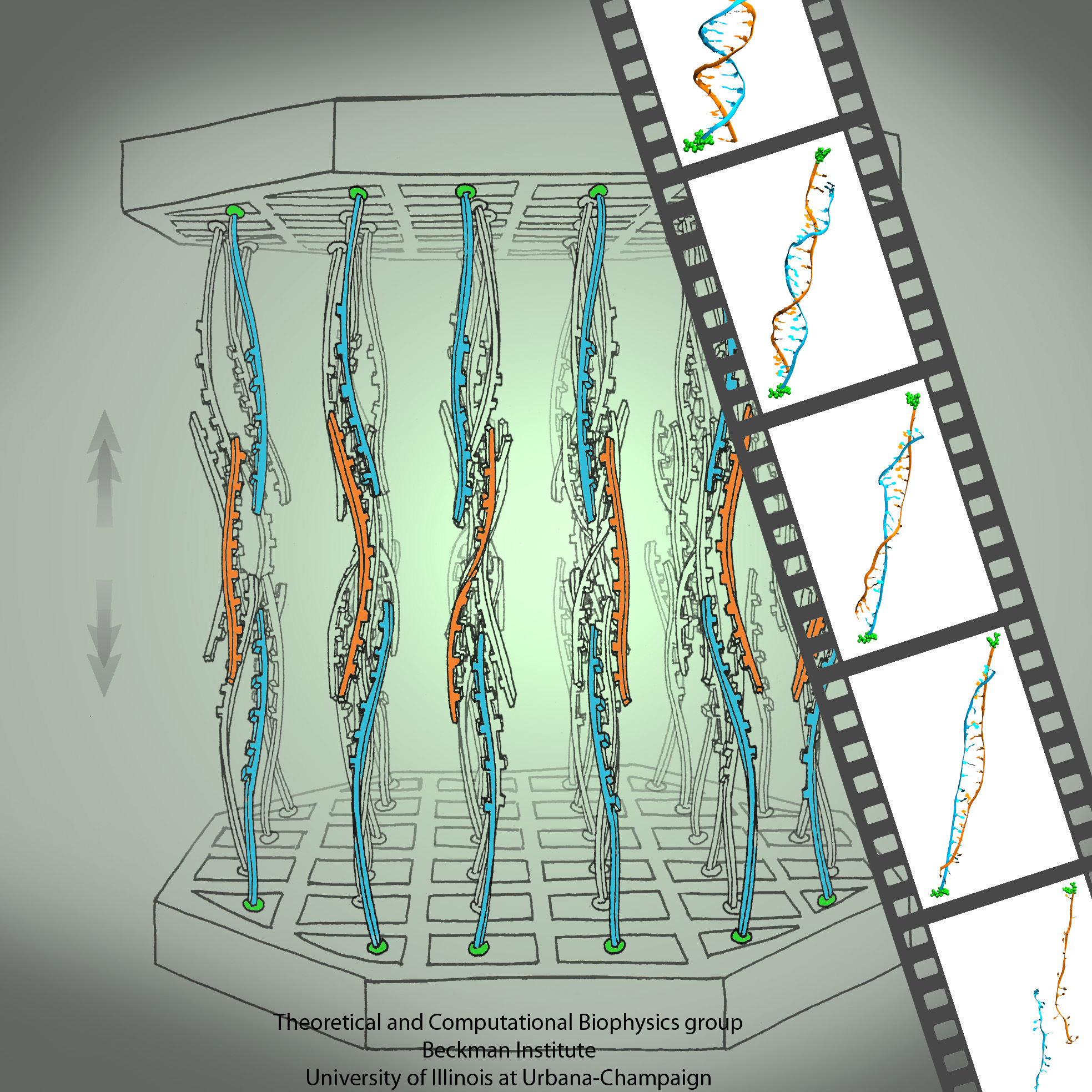
The genes of organisms, like plants and animals, offer the blueprint, not only to build the organism anew from a seed or fertilized egg cell, but also to adapt the living organism to its habitat and life experience. For example, a type of tree growing in an arid or wet region will adapt expression of its genes for root growth optimal to circumstances. A child living on a scarce or abundant diet or with little or much physical activity will adapt body growth accordingly. The needed, life time adaptation requires an individual's change in gene expression. This change is the subject of epigenetics. One control element in epigenetics is that cytosine bases of an organism's DNA become methylated in a chemical reaction in which a hydrogen atom is replaced by a methyl group (CH3). Another control element involves hydroxymethylation of cytosine bases where the H atom is replaced by a hydroxymethyl group (CH2OH); hydroxymethylation arises mainly in brain tissue. DNA methylation and hydroxymethylation patterns depend on an organism's individual history; aberrant patterns can be the cause of diseases, for example, of certain cancers. It is known that the proteins involved in gene expression can recognize methylated sites of DNA and, thereby, direct gene expression; DNA methylation also affects the packing of DNA in the chromosomes. However, methylation and hydroxymethylation may also affect gene expression directly; indeed, experiment and computational modeling with NAMD suggest this as an intriguing third way for methylation and hydroxymethylation to regulate gene expression. Methylation is shown, as reported recently, to make it more difficult to separate the two strands of DNA, as is necessary during gene expression. An earlier experimental-computational study had revealed already that methylated DNA can pass narrow synthetic nanopores more readily than unmethylated DNA can (see the Feb 2009 highlight). A recent experimental-computational study has shown that also in the case of hydroxymethylation mechanical properties of DNA become altered, namely, the forces needed to separate the two strands of DNA are strongly affected. The three experimental-computational findings advance our understanding of methylation and hydroxymethylation-based epigenetics and of how our body adapts to our life style and environment. More on our DNA methylation and hydroxymethylation website.
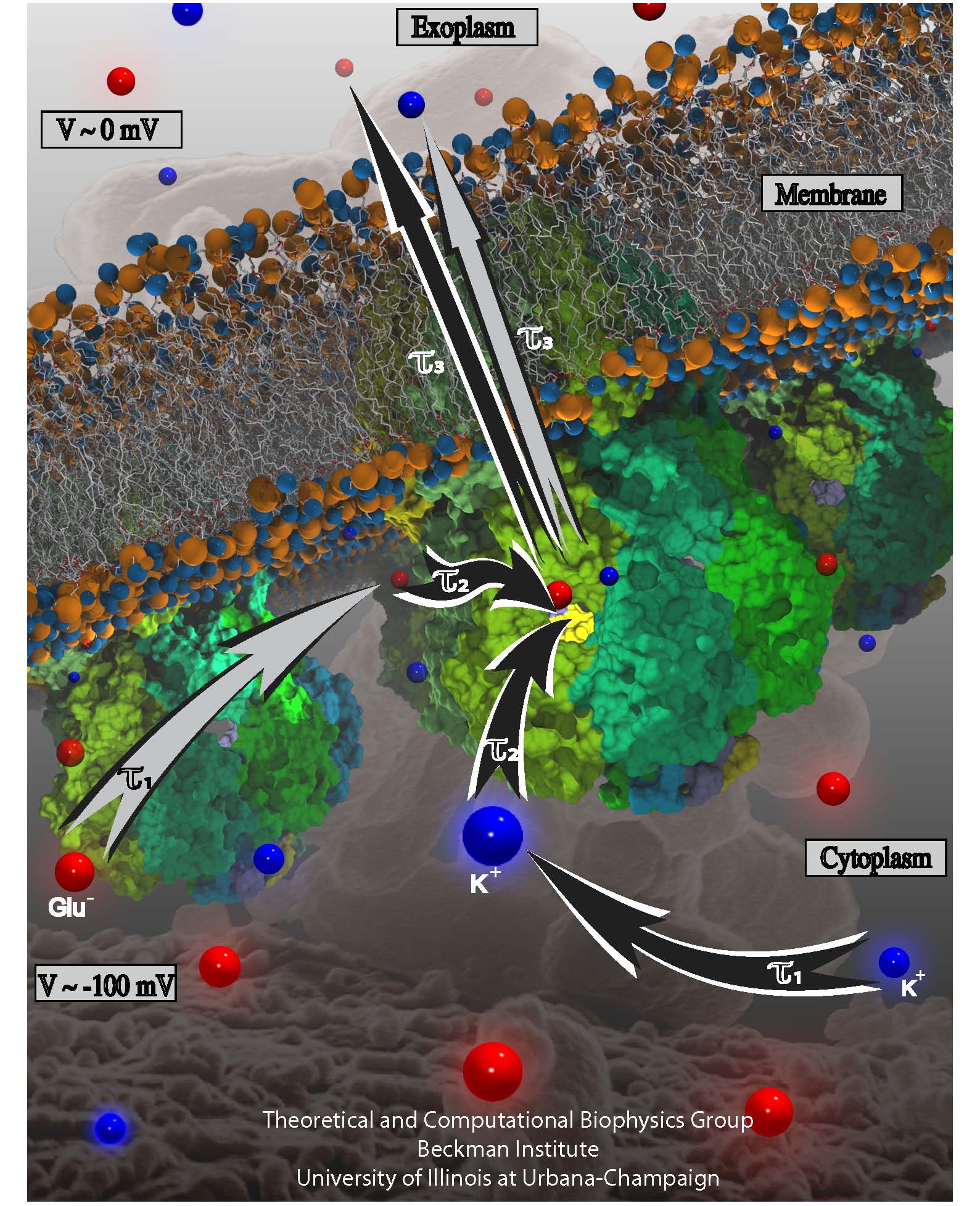
Bacterial cells enclose themselves with a cell membrane to maintain an optimal interior ion concentration and interior electrical potential. The potential is about -0.1 V inside versus 0 V outside, the potential difference amounting to an important energy reservoir for the cell. Closing itself off from the outside can be dangerous, though, for a cell, namely when sudden changes outside the cell can bring a cell to burst. This can happen when ions outside the cell are washed away suddenly: outside water pushes itself then into the cell as the water prefers to be near ions found then only inside the cell, a behavior called osmosis. The osmotic push can be so hard that the cell can burst. To prevent such burst, cells evolved safety valves, one being called mechanosensitive channel of small conductance or MscS. Under pressure, the valve, i.e., MscS, opens and enough ions leave the cell to keep water from pushing in too hard (see the Mar 2008 highlight, "Observation and Simulation depict Cell's Safety Valve", Feb 2007 highlight, "Observing and Modeling a crucial Membrane Channel", May 2006 highlight, "Electrical Safety Valve", and the Nov 2004 highlight, "Japanese Lantern Protein"). But since the electrical potential inside is negative, mainly negative ions would leave the cell, discharging its potential and leaving the cell without energy. A theoretical and computational study, the latter carried out using NAMD, reports now that MscS developed apparently an ingenious solution: ions going through the MscS valve must pass a balloon-like filter that manages to mix positive and negative ions so that only a 1:1 mixture leaves a cell under osmotic shock, thereby providing protection against the shock without compromising the cell's electrical potential. More information can be found on our MscS website.
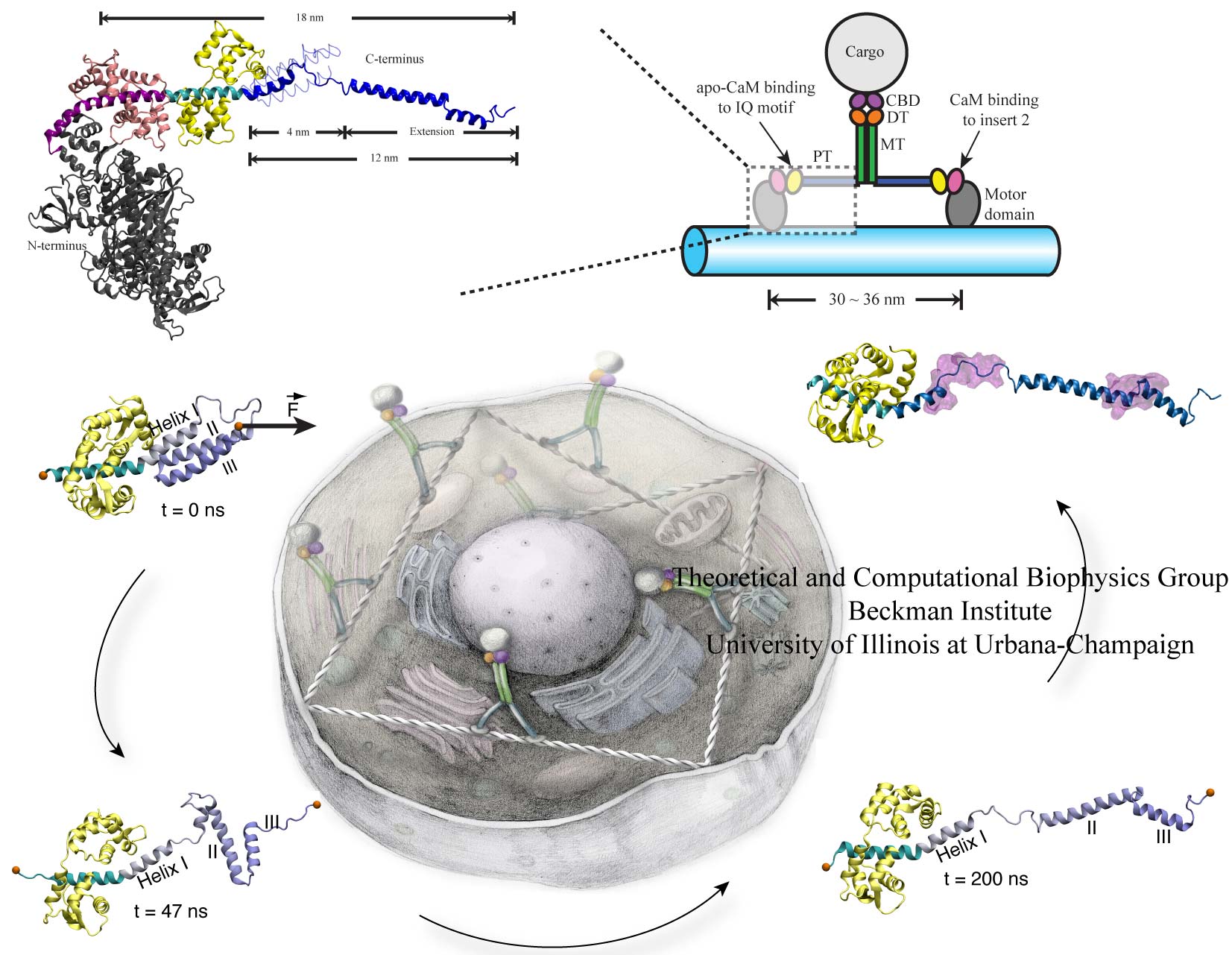
image size:
266.3KB
Motor proteins transport cargos from one place in living cells to another, for example transport cell components along the long axons of nerve cells. A motor protein, such as myosin VI, has to "walk" or "run" along the cellular highway of actin filaments to perform the transport. In the case of myosin VI, snapshots from crystallography revealed that the protein's "legs" are too short to explain the step size taken. Computational and experimental biophysicists have now solved the mystery of how myosin VI dimers realize their large step size despite their short legs. The investigation, based on the program NAMD and reported recently, demonstrates that the answer lies in the flexibility of the legs. Myosin VI is able to triple the length of each leg, made of a short bundle of up-down-up connected alpha-helices, by extending the bundle to a stretched-out down-down-down geometry of segments, like turning a letter z into a a single long line. In the telescoping process, myosin VI also gets help from its well-known binding partners, namely calmodulins. The calmodulins direct the telescoping of the protein legs as well as strengthen the extended legs. Together with an earlier study of the "neck" region of the molecule (see December 2010 highlight on Opposites Attract in a Motor Protein), the scientists have established how walking myosin VI achieves its wide stride. More information can be found on our motor protein website.
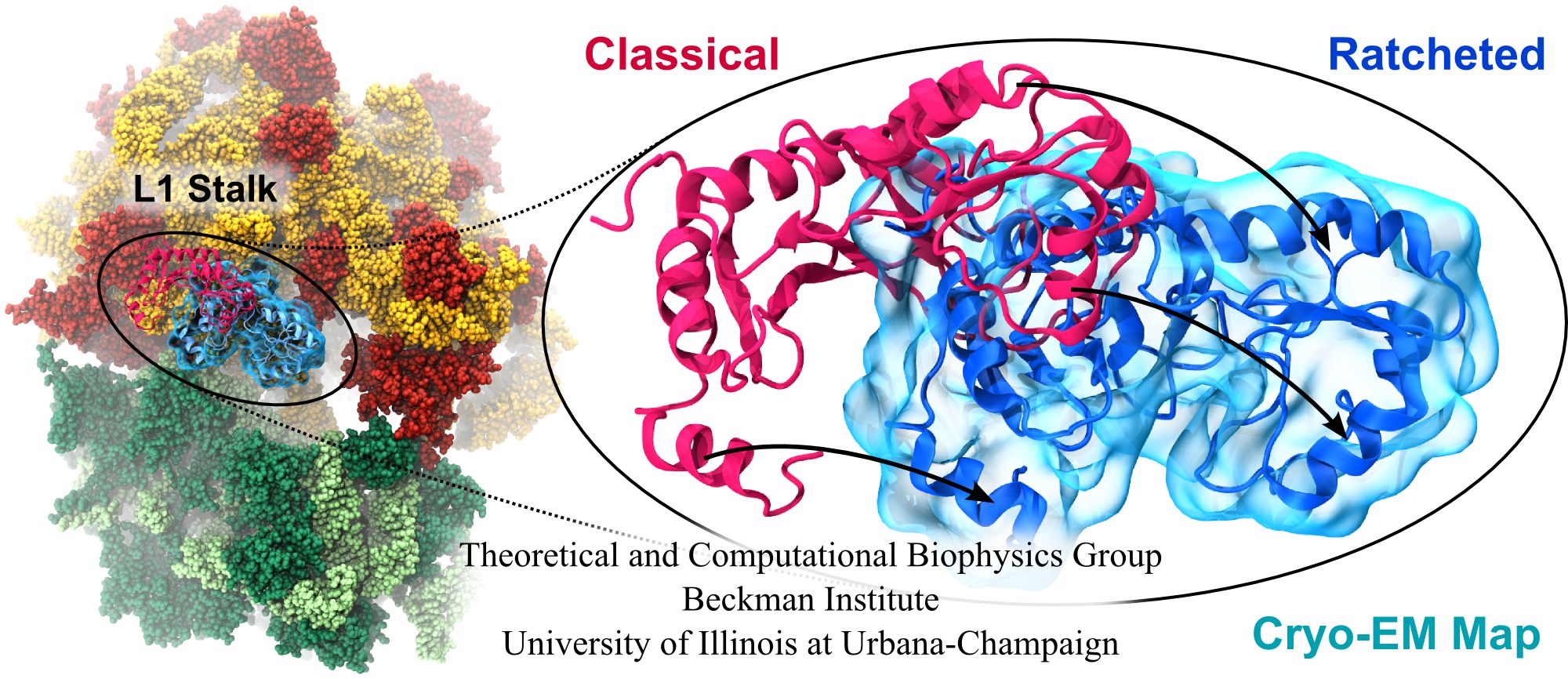
image size:
347.6KB
made with VMD
Water is the essential solvent that shapes protein structure and function, but for researchers using molecular dynamics flexible fitting (MDFF) to fit large biomolecular models to data from cryo-electron microscopy, such as fitting the classical ribosome into the ratcheted map, it was a mixed blessing. Since the network of hydrogen bonds that gives water its unique properties must rearrange as the solute moves, water molecules not only increase the size of the simulation but slow the fitting process. Leaving water out completely was a common practice, relying on the MDFF fitting potential to prevent the dehydrated protein from shriveling. The 2.8 release of NAMD provides a better option: a new implementation of the generalized Born implicit solvent model that scales to thousands of cores for large biomolecular aggregates thanks to NAMD's unique parallel structure and measurement-based load balancing system. By eliminating explicit water molecules from the simulation, an implicit solvent model helps shape protein structure while adapting immediately to new conformations. With this best-of-both-worlds option now available in NAMD, biomedical researchers using MDFF need fear water no longer.
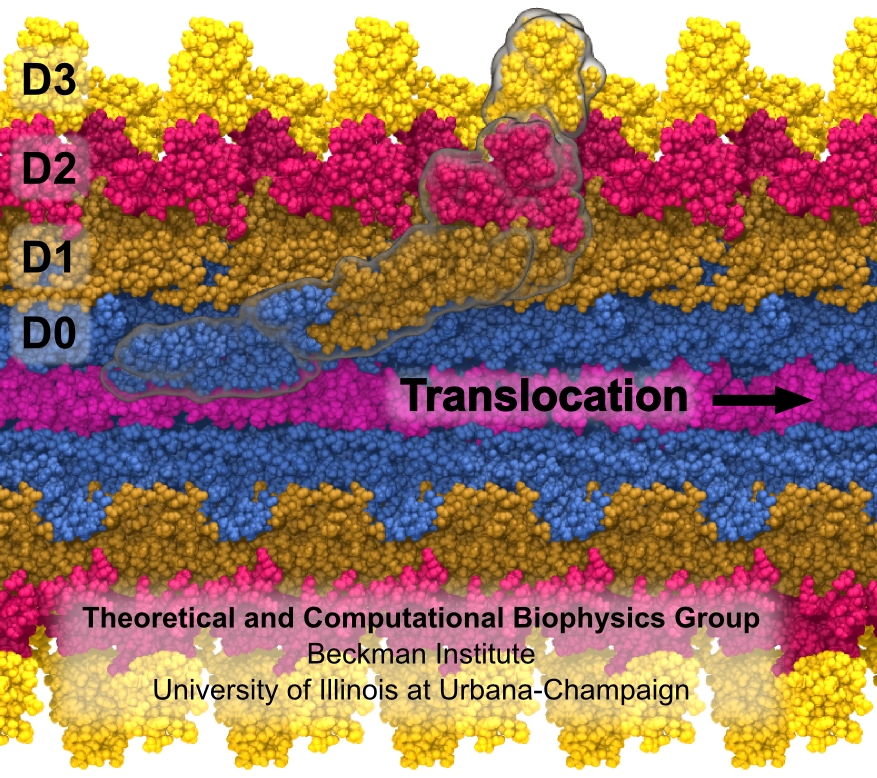
Bacterial cells can swim and use for this purpose one or more flagella, whiplike appendages that exceed the length of the cell severalfold. The flagella are made of many thousand copies of a protein called flagellin, arranged in a helical fashion such that the flagella are hollow inside, forming a very long channel. When the flagella are rotated by the cell counter-clockwise, the cell swims straight; when they are rotated clockwise, the cell turns to a new direction. Through swimming and turning the cell searches its habitat for food and avoids trouble. But sometimes a flagellum breaks and needs to grow back. At this point starts an amazing process: the cell makes new flagellin and pumps the unfolded protein into the flagellar channel, extending its length. This is like squeezing toothpaste out of a tube, except in reverse, like pumping toothpaste into the tube at the toothpaste factory, and the tube is extremely long. Now researchers have described the process that makes flagella grow step-by-step through a combination of mathematics, physics, and molecular modeling using NAMD. As reported, the researchers reproduce the time course of growth as well as the length of the growth and also explain how friction of the protein paste is kept extremely low to make the flagella grow many times the length of the cell itself. More information here.

image size:
1.9MB
Olga Svinarski and VMD
The molecular graphics program VMD has evolved to version 1.9, featuring new and improved tools for molecular cell biology. A key new tool in VMD permits visual diagnosis of the long-time dynamics of large structures. Improved computer power permits now simulation of processes like protein folding that stretch over microseconds to milliseconds. While short in human time, the process measured in terms of basic molecular motion like bond vibration is seemingly endless, involving hundreds of gigabytes of data and long hours of human attention, if not automated. VMD offers now with the Timeline tool, a convenient way of scanning such data for key "events" that signal successful process steps. Timeline can likewise assist in monitoring the dynamics of large cellular machines involving millions of atoms. VMD 1.9 sports other new tools like ParseFEP for analyzing so-called free energy perturbation calculations determining the energy arising in reaction processes as calculated with VMD's sister program NAMD. VMD includes now also a lightening fast tool to detect spatial regularities in the arrangement of small molecules in order to detect if they constitute disordered or ordered arrangements. By calculating the so-called radial distribution function the tool could identify plug formation in nanopore sensors due to precipitates that go undetected by density or visual inspection. Pleasing to the eye, molecular graphics features in VMD have been enhanced, enabling faster ray tracing, new shading features, X3D molecular scene export for display in WebGL-capable browsers including Chrome, Firefox, and Safari. See more on the VMD 1.9 release page.
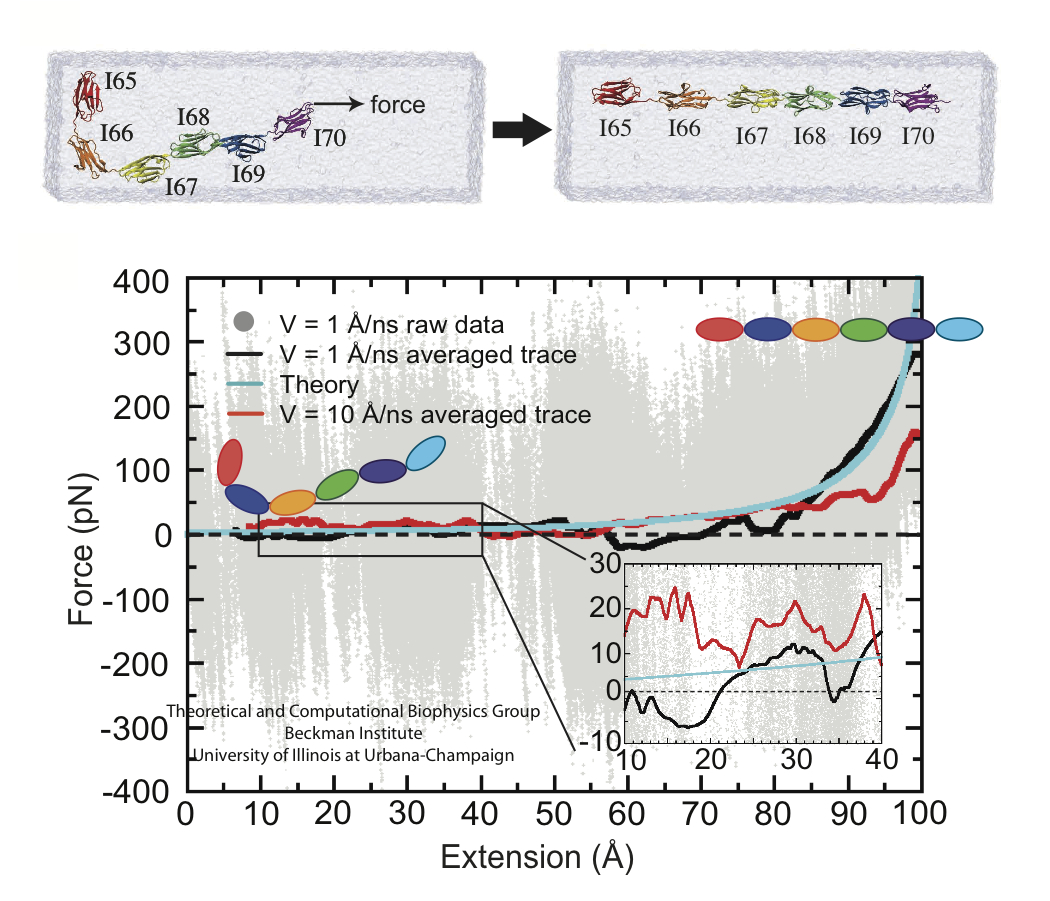
image size:
60.3KB
made with VMD
A cell that is alive can be recognized typically under a microscope through cell motion, streaming of inner cell fluids, and many other mechanical cell activities. These activities are the result of mechanical forces that arise in the cell, but even though the effect of the forces on cells are tremendous, the magnitude of the forces when measured by physical instruments, are extremely small, 1/1000000000000 of the force felt when lifting a Kilogramm. Sensing such small force requires extremely high precision in research techniques, and, for more than a decade, computational biologists, using NAMD, have exploited the all-atom resolution of molecular dynamics modeling to explain very successfully the molecular level effect of small forces in cells (for example, see our previous highlights on muscle protein, blood clot protein, and other successful applications). But simulations, too, are challenged by the small magnitude of cellular forces, mainly because precise simulation requires extremely extensive simulations, that cover a duration as close as possible to the biological time scale. Advances in computational biology have now permitted in the case of a the muscle protein titin a simulation of unprecedented accuracy that resolved clearly the relationship between molecular structure of titin and its ability to sustain the forces that arise in muscle function. For this purpose a key element of titin was stretched at a velocity slow enough that hydrodynamic drag, that came about as an unwanted byproduct of earlier simulations, was negligible compared to titin's intrinsic force bearing properties. The new simulations opened an unveiled view on muscle elasticity. More information can be found on our titin website, and in a recent review .

image size:
118.5KB
made with VMD
movie (YouTube)
Molecular modeling simulates the motion of cellular biomolecules at the atomic level. To make the simulations faithful, the physical forces acting between atoms need to be described accurately. Electric field effects between atoms, so-called atomic polarizabilities, are especially difficult to model well in a computationally cost effective way. There is an ongoing effort in the molecular modeling community to develop cost effective models that more faithfully represent the microscopic properties of biomolecules due to the ambient electric field effects. Recent development work has added support in the simulation program NAMD for one of these advanced modeling efforts. As reported, the new algorithms used in NAMD achieve good parallel computing performance, with a cost that is not more than twice that of the standard model, not accounting for atomic polarizabilities. The new model is demonstrated to reproduce many physical properties better than the standard model, including more accurate bulk density and surface tension at the interface between liquids and more accurate diffusive behavior of ions in a solution. More on our research webpage.
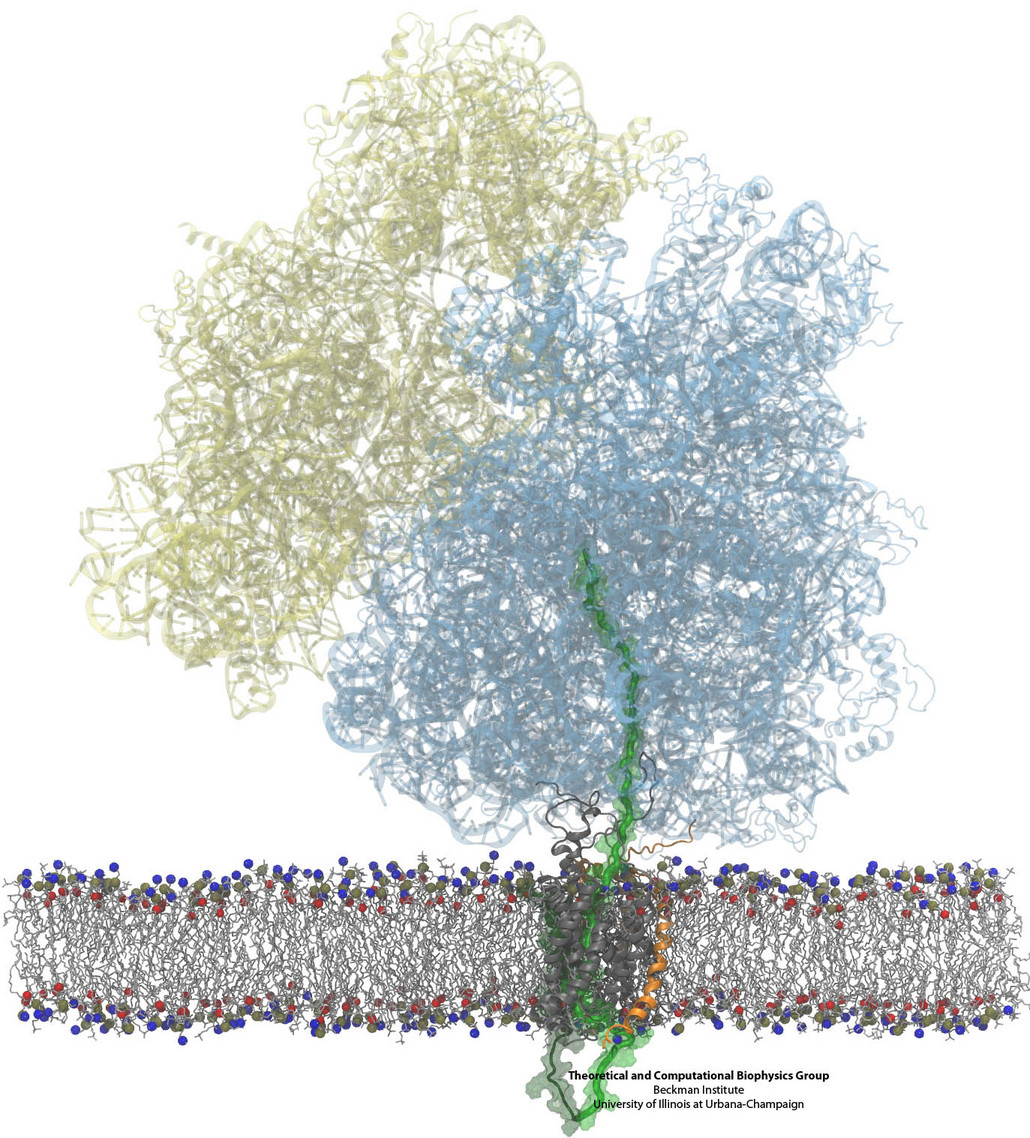
Constructing and correctly placing new proteins is a complicated task for living cells. Starting with nothing more than a sequence of DNA, the cell has to translate the genetic code, stitch together the constituent amino acids, and then place the newly made protein where its function is needed. To accomplish these feats, the cell using tools such as the ribosome, a protein factory (see the Dec. 2009 highlight Managing the Protein Assembly Line), and the protein-conducting channel, a switching station within the membrane (see the Nov. 2008 highlight Patching a Leaky Channel). All instructions for making a nascent protein and localizing it, e.g., to the watery cytoplasm or the oily membrane, are carried within its DNA sequence, but how it is read and then executed has long been unclear. Now, two recent research advances picture these processes in astonishing detail. The first advance (reported here), from a collaboration between cryo-electron microscopists and computational biologists using MDFF, shows an atomic level structure that caught the ribosome in the act of inserting a protein into a membrane. The structure reveals the newly forming protein transiting from within the ribosome to the channel and then through an open gate in the protein-conducting channel into the membrane. The second advance (reported here), accomplished with the simulation program NAMD, explained how the ribosome and the protein-conducting channel manage to pay the energy price of inserting a new protein, one amino acid a time, into the membrane. For more information see our protein translocation website.
A recent review explores how the Graphics Processing Units (GPUs) found in commodity high-end video cards are increasingly being used not only for interactive molecular graphics, but also for molecular simulation and analysis. NAMD and VMD both support GPU-acceleration using NVIDIA CUDA, enabling computationally demanding simulation, visualization and analysis tasks (e.g., of the electrostatics of Tamiflu binding) to be run with shorter turnaround on modestly priced GPU clusters, desktop, and laptop computers. This affordable computational power is particularly compelling for interactive modeling, with a recent report detailing how interactive molecular dynamics simulations with haptic feedback are now possible on GPU-accelerated desktop computers. Three recent book chapters detail the application of GPU computing techniques to the calculation of electrostatic potentials, interactive display of molecular orbitals, and more general molecular modeling algorithms. In August 2010 the Resource held its first workshop on GPU programming for molecular modeling to bring the benefits of GPU computing to a broader range of molecular modeling tools and magnify the impact of our GPU computing research.

image size:
86.3KB
made with VMD
Motor proteins are fascinating cellular machines that convert chemical energy into mechanical work. They are employed in a wide range of cellular functions like muscular contraction, transportation of proteins and vesicles, and cell motility. Myosin VI is an example of a motor protein. It "walks" along actin filaments (kind of like cellular highways), performing tasks such as delivering materials across the cell. Primarily, myosin VI functions as a dimer (i.e., two myosin VI proteins are associated and form a functional complex), but the structure of the myosin VI dimer, particularly how a myosin VI associates with another one, is still debated. Teaming up with experimentalists, computational biologists investigated how two myosin VI assemble and pull their cargo together. The investigation, reported recently, focused on a segment of myosin VI that forms a long, rigid alpha-helix that is notably decorated with a distinct rings of positively and negatively charged amino acids. Carrying out single-molecule experiments along with molecular dynamics simulations using NAMD, it was found out that two myosin VI proteins attract each other electrostatically through the charge-ring proteins, shifting them such that the oppositely charged amino acids from different helices face each other. More information can be found on our motor protein website.
image size:
994.9KB
made with VMD
The eagerly awaited 2.7 release of NAMD includes GPU acceleration, downloadable binaries for Linux clusters with InfiniBand networks, grid-based forces and extra bond/angle/torsion conformational restraints for molecular dynamics flexible fitting (MDFF), and a major enhancement of both alchemical and conformational free energy methods. Alchemical calculations gradually alter the chemical structure of a molecule during a simulation, monitoring the consequences of creating and deleting atoms via either free energy perturbation or thermodynamic integration. A soft-core correction may be employed to circumvent singularities when atoms are created. Conformational calculations can probe structural rearrangements along an arbitrary number of collective variables, including distances between atoms or groups of atoms, distances projected along an axis or in a plane, angles, torsions, eigenvectors, gyration radii, coordination, root mean-square displacements, orientations, and alpha-helicity. New variables can be introduced without recompilation. Free energy surfaces or potentials of mean force can be constructed using metadynamics, the adaptive biasing force method, umbrella sampling, and steered molecular dynamics. The collective variables capability of NAMD has allowed the simulation of the illustrated hexameric helicase. In the simulation, a single strand of DNA passes through the central core while the hexamer translocates or "walks" along the DNA, a mechanical process driven by the energy of ATP hydrolysis.
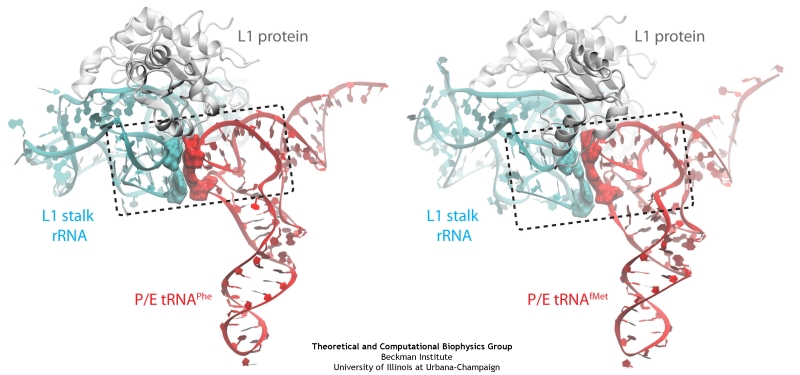
image size:
140.2KB
made with VMD
The ribosome is a molecular machine ubiquitous in all living cells and translates genetic information into proteins. Proteins are made of twenty different amino acids, strung in a linear sequence. The amino acids are coded for by the genes in DNA, but for the purpose of protein synthesis genes are transcribed into a working copy, a messenger RNA. The latter is translated by the ribosome into proteins with the help of transfer RNAs, which bring the individual amino acids. There is a transfer RNA for each of the twenty amino acids. Much progress has been made regarding the static structure of the ribosome, transfer RNA, and nascent protein components (see also the Dec 2009 and Jan 2009 highlights Managing the Protein Assembly Line and Open Sesame). Now researchers are looking into the inner workings of the whole system combining various experiments and computational modeling using NAMD and VMD. The combination yielded unprecedented detailed views of the ribosome in action as reported recently, namely how a dynamic part of the ribosome helps guiding transfer RNAs on their way out of the ribosome, and explains why transfer RNAs behave differently on their journey, depending on if they start synthesis of a protein or if they elongate a protein. More on our ribosome website.
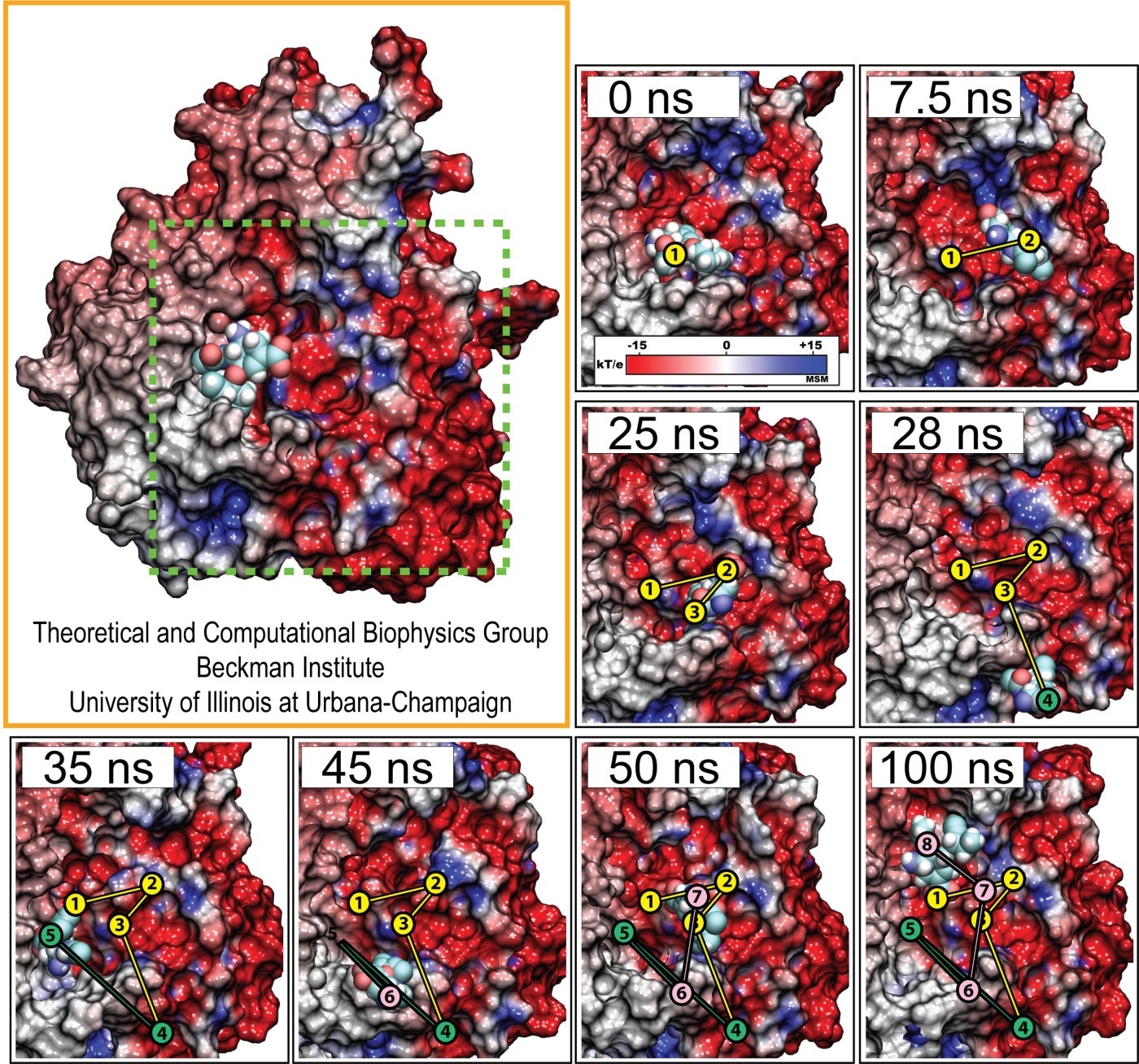
Fever, chills, sore throat, coughing, aches, and pains? Ah ..... you have the flu! As a measure of prevention, vaccines against seasonal influenza are distributed and administered each fall. Last year though, the outbreak of the H1N1pdm "swine" influenza virus, caught health workers by surprise as this virus not only infected individuals during the spring and summer months, but also seemed to be particularly virulent towards otherwise healthy young people. Even more alarming was increasing evidence that H1N1pdm had acquired resistance to the frontline antiflu drug, Tamiflu. In response to this, computational biologists at the University of Illinois and the University of Utah teamed up to uncover the basis for influenza drug resistance through quantum chemistry, and molecular dynamics simulations with NAMD. The results of this study have recently been reported, and uncovered a two stage binding pathway for Tamiflu in H1N1pdm "swine" and H5N1 "avian" flu proteins, as well as a possible mechanism through which genetic mutations can induce drug resistance in one of the stages. Subsequent efforts at drug design against influenza can take advantage of this discovery. This discovery was made possible through use of so-called GPU computing (see Oct 2007 highlight "Graphics Processors Speed Up Simulations"). More information can be found here.
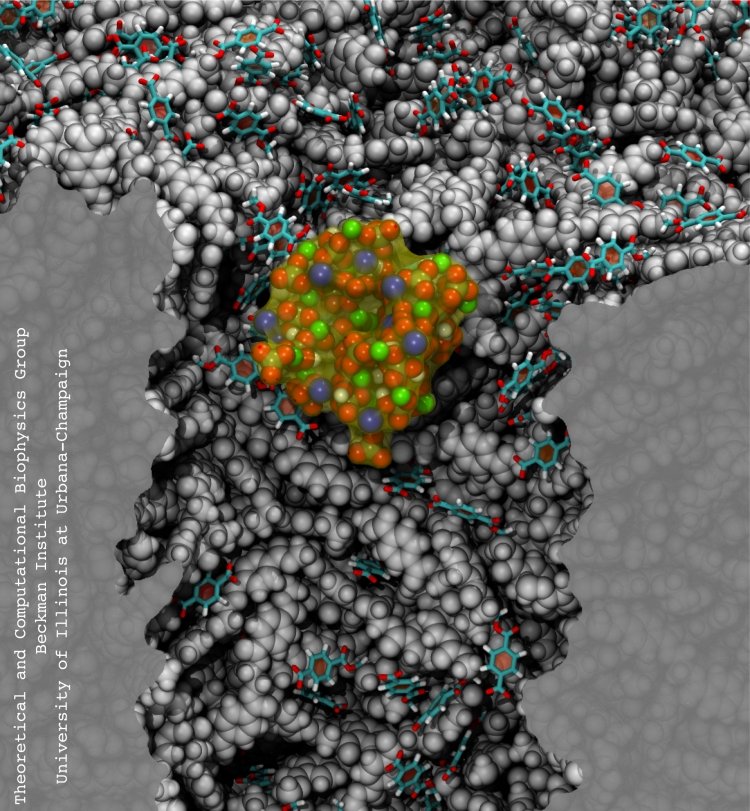
Nanopores are promising new sensors, which may soon sequence DNA in a cheap and fast way (see Nov 2005 and Oct 2004 highlights) and control the flow of ions in nanodevices (see Dec 2009 and Mar 2009 highlights). In measurements, DNA molecules and ions pass through the nanopores which are only a nanometer (0.000001 mm) wide. Unfortunately, researchers have to battle clogging of the very narrow nanopores. For example, it was found that small concentrations of so-called divalent cations produce clogging of plastic nanopores. To figure out how to prevent clogging, researchers needed a microscopic view of the process. Such view was furnished through molecular dynamics simulations, based on NAMD, of ion conducting nanopores. In a recent study, an atomic-level view of the clogging process, revealing the formation of tiny ionic crystals inside the nanopore, was offered. The view suggests a remedy against nanopore clogging. More information can be found in our polymeric nanopore website.
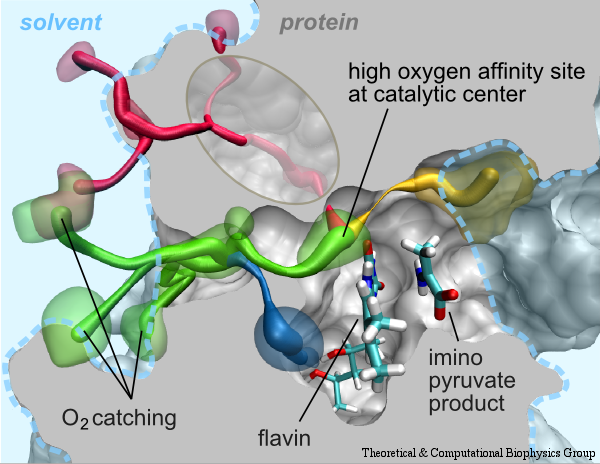
image size:
225.2KB
made with VMD
Cells contain numerous enzymes that use molecular oxygen for their reactions. Often, their active sites are buried deeply inside the protein, which raises the question whether there are specific access channels guiding oxygen to the site of catalysis. Localizing oxygen molecules in proteins is difficult. Oxygen is very mobile and difficult to locate by most experimental techniques. However, computer modeling can come to the rescue. In a recent study computational biologists using NAMD could identify the D-amino acid oxidase regions where oxygen molecules most likely reside. D-amino acid oxidase is considered essential for understanding catalysts that involve flavin moieties in proteins. The reported findings reveal a channel system through which oxygen molecules diffuse from the protein surface to the protein's catalytic center. Indeed the channel ends at a site in the protein that constitutes the ideal location for oxygen to react with flavin. The computational results are reported to be verified through so-called site directed mutagenesis experiments. Click here to read more.
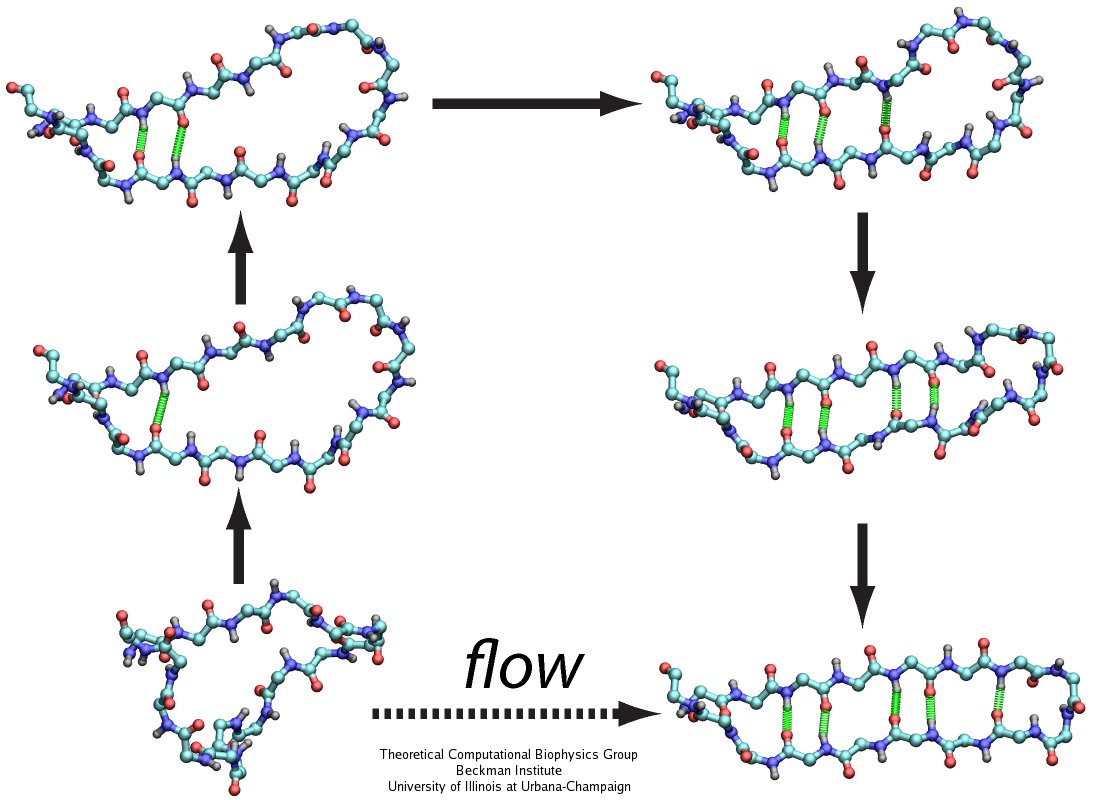
image size:
110.0KB
made with VMD
The human body is protected by self-healing mechanisms, one of them being instant blood clotting at a bleeding site after blood vessel injury. What triggers the formation of a blood clot? Researchers found that a protein on blood platelets, called GPIbα, functions as a sensor of so-called high shear flow caused by bleeding. A loop-shaped, 17-amino-acid-long, segment of GPIb&alpha, the β-switch, acts as the flow sensor. Once a blood vessel is injured, bleeding increases shear stress due to blood flow at the wound, which in turn induces the β-switch to change from a loose, loop-shape to an elongated, hairpin-shape, the latter referred to by researchers as a β-hairpin. This conformational change makes GPIbα stick better to the damaged vessel and eventually leads to blood clotting, which heals the vessel. In a prior study (see the Jul 2008 highlight, Molecular Flow Sensor Triggers Wound Healing), Molecular dynamics simulations using NAMD and VMD provided already a microscopic view of the flow-induced loop to β-hairpin transition. A recent study extended the investigation of the remarkable biological flow sensor, detailing the flow rate needed to trigger it and identifying the detailed sensor mechanism. A combination of simulation and mathematical analysis revealed the β-switch as a system of two stable states, one disordered, with loop geometry and one ordered, with β-hairpin geometry. Normal flow prefers the disordered state; high shear flow prefers the ordered state, inducing thereby the life saving transition. More on our flow sensor website.
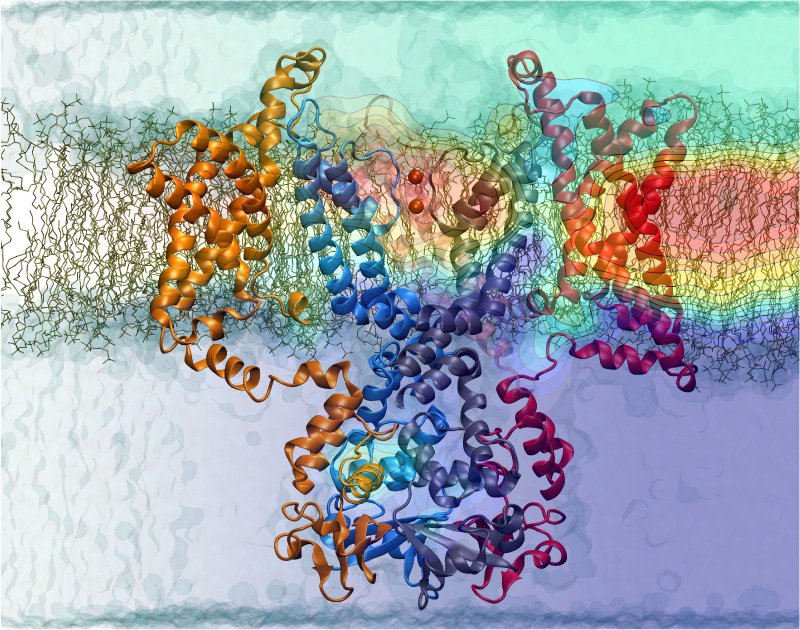
Nerve cells, through their electrical signals, control actions and intelligence of higher organisms. The signals result mainly from potassium and sodium ion channels in the cells: when the cells are stimulated electrically, they send an all (in case of sufficient stimulation) or nothing signal to other nerve cells or organs like muscle. As shown in ground breaking work by 1963 Nobelists Hodgkin and Huxley, cast into mathematical equations, nerve cells establish these signals through voltage gating of channels. The nature of the gating, monitored through the so-called gating current, has been elusive for decades, despite a detailed characterization of the ion conductivity itself rewarded through a 2003 Nobel Prize to MacKinnon. The riddle is that the channel involves a protein with few charged amino acids that seem to be only weakly coupled energetically to an electrical potential gradient across the cell membrane. Now a sweeping modeling study using NAMD employing the most powerful computers available to researchers today has led to an explanation of voltage-gating. Simulations revealed that the potential gradient is focused by the channel protein to a very narrow region such that its value is much larger than anticipated. The protein was also seen to arrange its charged amino acids sensing the gradient in an unusual helix, a so-called 310 helix, that aligns charges perfectly while at the same time inducing a motion that opens and closes the channel. Proof of the veracity of the computational model is that the calculated gating current fits perfectly the observation. More on our potassium channel website.
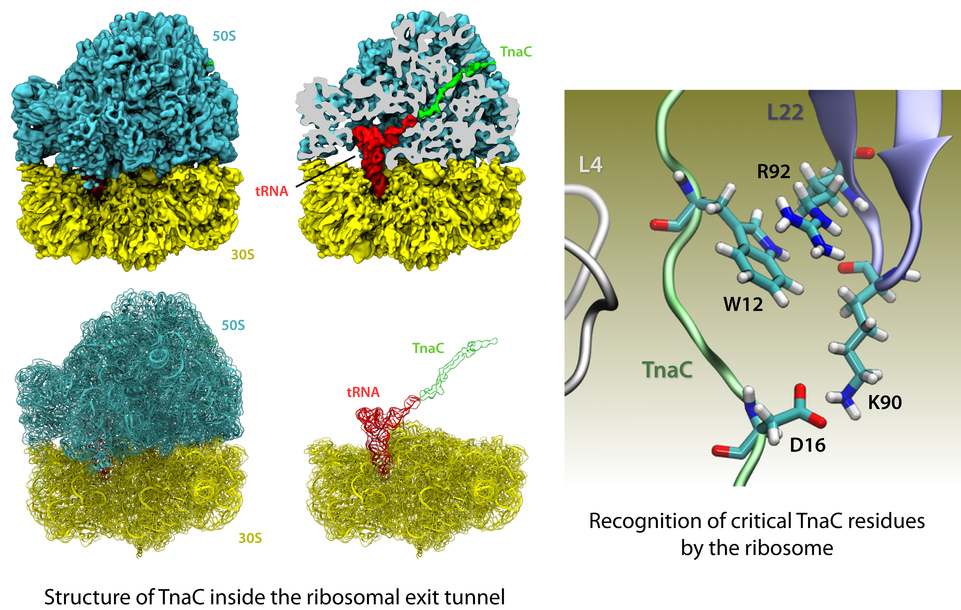
image size: 541.6KB
The ribosome is the protein factory of all cells. While proteins are being synthesized, they must travel through a tunnel inside the ribosome before they reach their required location in the cell. This tunnel recognizes certain sequences of nascent proteins and responds to them in various ways. For example, certain protein sequences recognized in the tunnel can shut the ribosome down, stopping protein synthesis. A much studied example is the bacterial protein TnaC, which by shutting down its own synthesis turns on the synthesis of proteins involved in the degradation of a molecule called tryptophan. The structure of TnaC inside the ribosome was previously determined through crystallography, electron microscopy, and computer modeling (see the December 2009 highlight on Managing the Protein Assembly Line). Simulation of TnaC dynamics inside the ribosome using NAMD and VMD has now revealed the mechanism by which TnaC shuts down the ribosome as reported recently. Answered was also the question of how the ribosome recognizes the sequence of TnaC and why many proteins that have sequences similar, but not identical, to that of TnaC do not shut down the ribosome. For more details, see our ribosome website.
Harvard University and Harvard Medical School News Alert: Our senses are essential for survival and for the exploration of natural environments, and much has been learned about the molecular basis of vision, olfaction, and taste. Yet only a few of the molecules mediating touch and sound perception have been discovered. Now, a team from MCB and HMS has resolved the molecular structure of cadherin-23, a key protein important for sound perception. They have used this structure together with molecular dynamics simulations to understand its mechanics and its function in hearing and deafness.
With the cadherin-23 structure in hand, the team used molecular dynamics simulations to determine its elasticity. The tip link has been assumed to be a relatively elastic, spring-like molecule. However, an extensive set of atomistic simulations, performed using parallel supercomputers with thousands of processors and the NAMD software developed at UIUC, suggest otherwise. The simulations revealed a stiff cadherin-23 molecule, with tightly-bound calcium ions preventing mechanical unfolding. The tip link is therefore suggested to be a stiff cable conveying force to transduction channels, with some undetermined molecular component providing the necessary elasticity for the system.
image size:
212.3KB
made with VMD
A smart strategy usually involves a plan B. As it turns out, the muscle proteins in our bodies responsible for the physical motions like running or the beating of our hearts, also rely in their function on having a plan B strategy. When contracting and extending, muscle fibers generate tremendous forces that need to be buffered to protect muscle from damage. This role falls to the muscle protein titin, which is composed of a chain of linked domains, making it a molecular rubber band. When a small force is applied, titin employs its plan A and stretches apart without unraveling its individual domains (like what the movie on the side shows). When a stronger force is applied, plan B kicks in and further elasticity is generated by the unwinding of the protein domains one at a time. By practicing two modes of response to different levels of forces, titin provides the elasticity that muscle needs at a minimal structural cost. A recent computational-theoretical investigation has provided a molecular view on how titin's two plans work, the study featured in a journal cover. The needed simulations were performed using NAMD. Principles described in this study can also be found in other mechanical proteins, recently reviewed here. More on our titin IG6 website.
image size:
115.2KB
made with VMD
Bacteria contain the simplest photosynthetic machineries found in nature. Higher organisms like algae and plants practice photosynthesis in a more elaborate but principally similar manner as bacteria. But even for its simplicity, the bacterial photosynthetic unit is not without its unsolved mysteries. Take, for example, the crucial photosynthetic core complex, which performs light absorption and the initial processing of the light energy. In certain bacterial species, the core complex contains two copies of an additional small protein (made of about 80 amino acids) called PufX, whose role in photosynthesis is still a puzzle, and its location within the core complex is yet to be pinpointed. Numerous imaging studies have been published, yielding two opinions on what the role of PufX is and where exactly it resides. One opinion assigns the protein the role of gate keeper, the other the role of coordinator. Recently, a computational investigation was carried out that much supports the second role. Since PufX comes as a pair, two copies of PufX were placed side-by-side in a biological membrane and they were seen to adhere to each other strongly, but assume with their cylindrical (helical) shape an angle of 38 degrees. This geometry is perfectly suited for PufX to join the two parts of the symmetrical core complex together in the middle and to impose on the parts the tilt that was actually observed in the imaging studies. The needed simulations were done with NAMD. More details can be found on our photosynthetic core complex website.
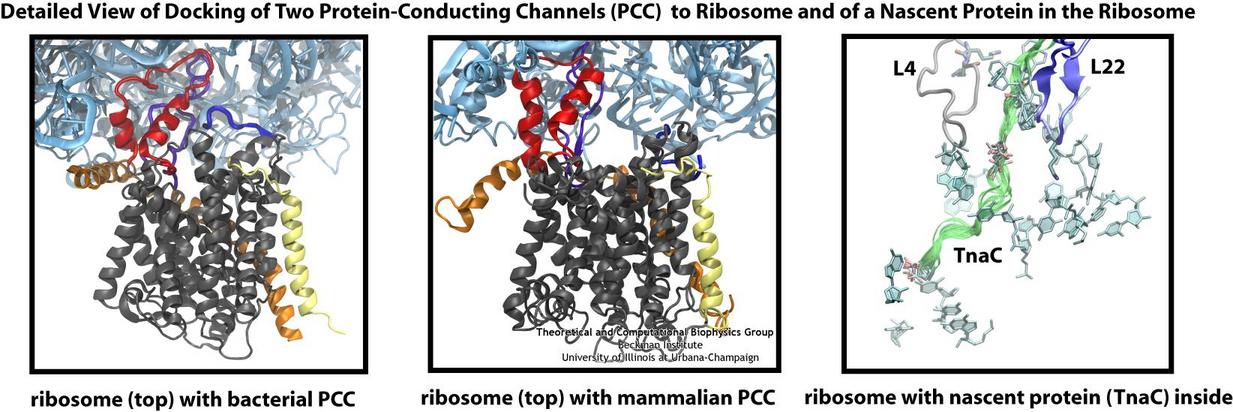
Living cells contain millions of proteins composed of sequences of different amino acids that typically fold spontaneously into well-defined three-dimensional conformations and then carry out their role as molecular machines serving manifold functions in the cells (see the protein folding highlight). The synthesis of the proteins is carried out by the ribosome, one of the largest molecular machines present in all cells, which reads the cell's genetic information for the purpose. Three researchers were recently awarded the 2009 Nobel Prize in Chemistry for the determination of the ribosome's structure. The physical mechanism of the ribosome, the cell's protein factory, is still largely unknown. Just as in any factory, there are multiple directions and controls on the protein assembly line. Sometimes the protein products need to be redirected to different parts of the cell, and other times assembly needs to be halted altogether. Now, significant new insights into both of these aspects of protein assembly have been made by combining electron microscopy with molecular dynamics simulations using the recently developed molecular dynamics flexible fitting method (MDFF, see the June 2008 highlight). This combination allowed researchers to visualize the complex between the ribosome and a protein-conducting channel that directs proteins into and across membranes for both a mammalian system (reported here) and a bacterial system (reported here). Amazingly, despite the evolutionary distance between mammals and bacteria, both complexes are remarkably similar. Simulations of the bacterial ribosome-channel complex, among the largest ever performed, further revealed the steps in the direction process. In a third study (reported here), researchers determined how TnaC, as a protein newly synthesized by the ribosome, can stall the ribosome from within during its own assembly, which then controls the expression of related genes. More information on these unique protein assembly controls can be found on our ribosome and our protein-conducting channel websites.
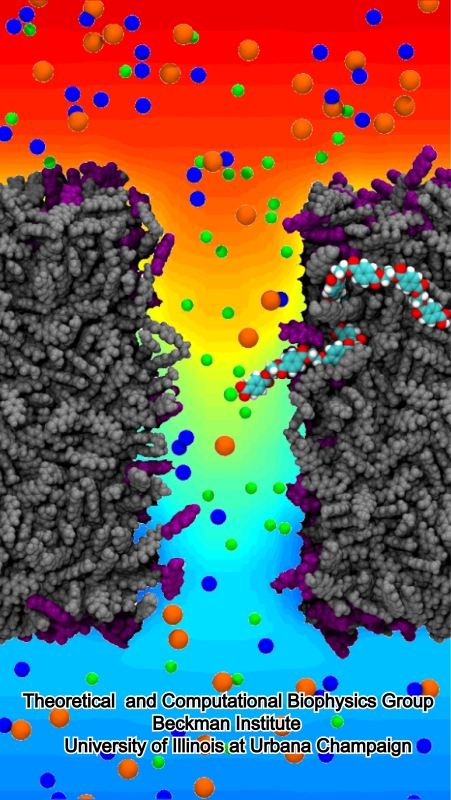
image size:
268.6KB
made with VMD
Nanotechnology is flourishing today, for example building entire chemistry laboratories on a small chip. In doing so nanoengineers experiment with different materials, one of the latest being good old plastic, chemically known as polyethylene terephthalate, or PET. Presently, nanometer-size pores, so-called nanopores, are built from silicates and PET membranes. To image how nanopores filter ions (see the Mar 2009 highlight) or affect DNA mechanics (see the Feb 2009 highlight) one can employ molecular dynamics (MD) simulations using NAMD. Such simulations provide a realistic representation of the devices, which can be employed to improve the technical uses of nanopores. In a recent publication, MD simulations revealed that the conduction properties of PET nanopores are controlled by chemical properties of the inner surfaces of the nanopores, for example, if surface groups sticking into the nanopore are protonated or not. The studies show once more the value of molecular dynamics simulations as computational microscopes, providing to nanoengineers the atomic-level visualization of extremely small devices. More information is available on our PET nanopore website.

image size: 493.4KB
Twenty years ago the Beckman Institute opened on the campus of the University of Illinois and the Theoretical and Computational Biophysics Group (TCBG) was the first to move into the beautiful new building. The group advanced computational biology through widely used software and exciting discoveries (some recently described here). Hence, it was time to celebrate and so TCBG did with the symposium Computational Biology of the Cell - The Next Decade. But rather than looking backward, the symposium glanced into the future that promises as many great opportunities as the last two decades realized: computational power for millisecond atomic resolution simulations, coarse-graining methods to describe true cellular size and times scales, better force fields, closer collaboration with experiment, and partnership with nanotechnology. Bringing together many of today's leading thinkers and practitioners of biomolecular modeling lectures led to lively debates (see some photos) regarding computational biology's future. On TCBG's past and future read more here.
Proteins are molecular machines inside living cells which carry out tasks from chemical synthesis to cellular motion. Each protein is a linear polymers made of units that each are one of twenty amino acids, A(1) to A(20), for example from a sequence A(5) - A(12) - A(1) - A(5) - ... - A(17). Random sequences lead to disordered, non-functional proteins. A central feature of life is that evolution has given rise to protein sequences which fold into functional proteins. The process through which the disordered linear polymer folds into its final structure, however, remains mysterious, since it is very difficult to observe experimentally. Unfortunately, simulations of protein folding are very computationally demanding and have uncertain outcomes (see the May 2009 highlight), which has historically limited their utility. Now, researchers have used NAMD to simulate the complete folding process of a small protein, villin headpiece, as reported recently. In a series of atomic resolution molecular dynamics simulations, covering a total of 50 microseconds, multiple folding events were observed. Importantly, the simulations provided a glimpse at the prevalent intermediate conformations visited by villin during folding. A key transition between two intermediates was recognized as rate-limiting in the villin folding process. More information is available at our protein folding website.
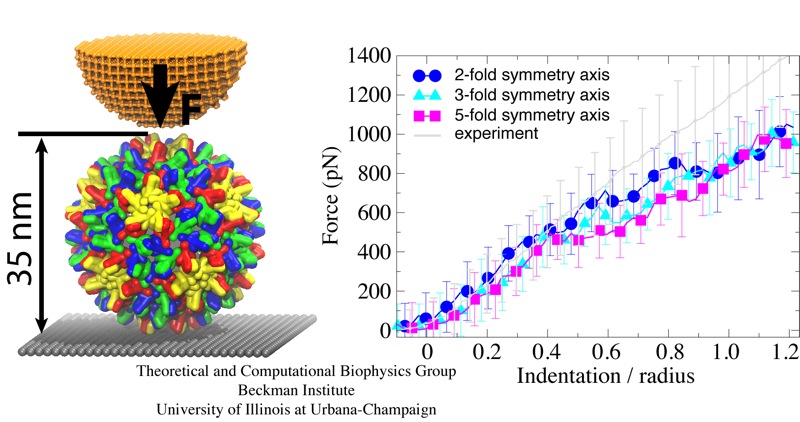
Viruses are the simplest life forms known. In fact, one can question if they are life forms at all, as they cannot exist without infecting a host cell and using its machinery for replication. The virus is indeed just a package material surrounding a genetic message that instructs the host cell to replicate the virus. It looks like a soccer ball, but is a million times smaller (see also the March 2006 and January 2007 highlights). The infection, a well known example being infection of human cells by a flu virus, involves the virus to approach a human cell and dock onto it, become internalized by the cell, bursting then its package, called the capsid, and release the genetic message. The virus capsid needs to be sturdy and impermeable up to the approach to the cell, but then become brittle and porous to release the genetic material. Obviously, the virus capsid must have very distinct mechanical properties to function. To investigate these properties experimental and computational biophysicists teamed up. The experimentalists placed empty capsids of the hepatitis B virus onto a small chip and mechanically squeezed the capsid then with an extremely small tip, measuring how much force is needed to squeeze the spherical capsid down repeatedly. Computational researchers using NAMD repeated the experiment in simulation. As they reported recently, simulation gave the same forces as the experiment, but yielded also a detailed picture of the capsid mechanics. More on our "Molecular dynamics of viruses" web site.
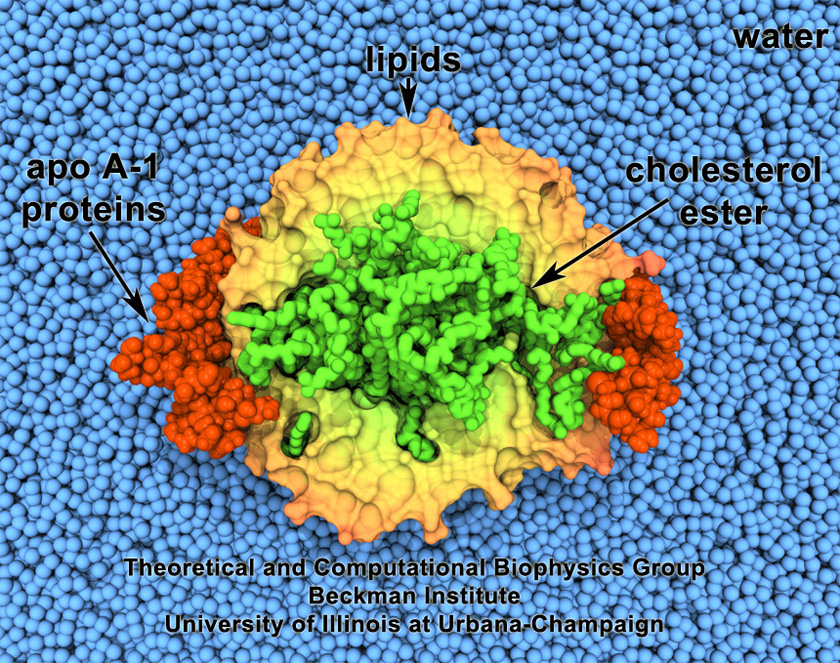
image size:
496.6KB
made with VMD
Cholesterol maintains a healthy body, but too much cholesterol can lead to atherosclerosis and heart disease. Lipoproteins can bag superfluous cholesterol in the arteries and transport it to the liver for removal. One such lipoprotein is high-density lipoprotein (HDL) which self-assembles into discoidal particles (see the Mar 2007 highlight) and then bags cholesterol. How this works is the subject of a recent report. Molecular dynamics simulations using NAMD revealed that discoidal HDL particles, teaming up with the enzyme LCAT, first turn cholesterol chemically into cholesterol ester and then sucks it into the interior of the particle; in the course of this process, the HDL particle swells into a sphere. More information on our lipoprotein website.
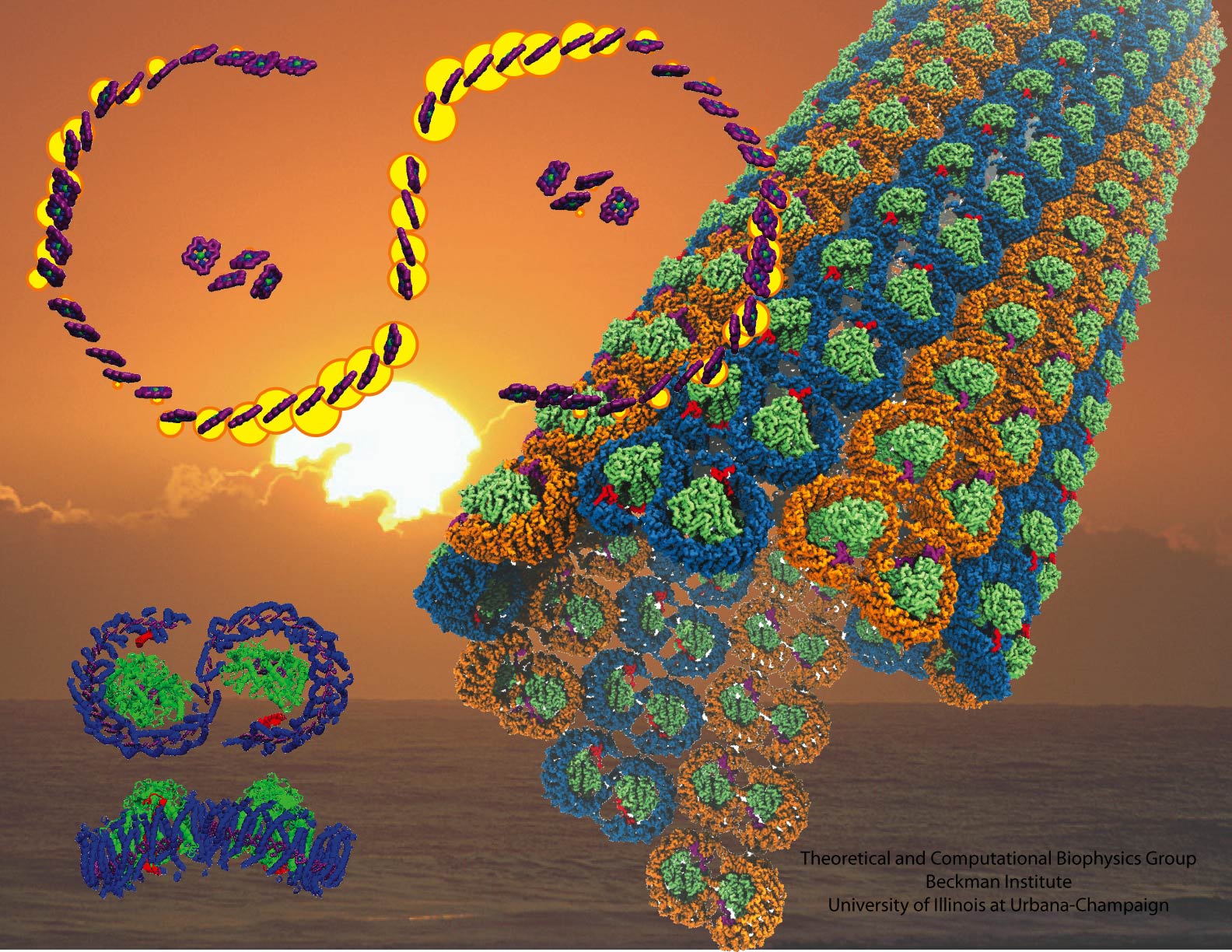
image size:
440.0KB
made with VMD
Proteins are the workers in cells; they carry out designated cellular functions tirelessly throughout their lifetimes. Some proteins can even hold two different jobs. One example of a dual-duty protein is the bacterial photosynthetic core complex. The photosyntehtic core complex performs the first steps of photosynthesis: absorption of sunlight and processing of light energy. Besides providing solar power, the core complex acts as an architect of the cell by shaping membranes in the interior of photosynthetic bacteria. Combining computational modeling and electron microscopy data using the Molecular Dynamics Flexible Fitting method, computational biologists have recently reported studies of both functions of the core complex, namely, the light-absorbing features and the membrane-sculpting properties. More details can be found on our photosynthetic chromatophore website.
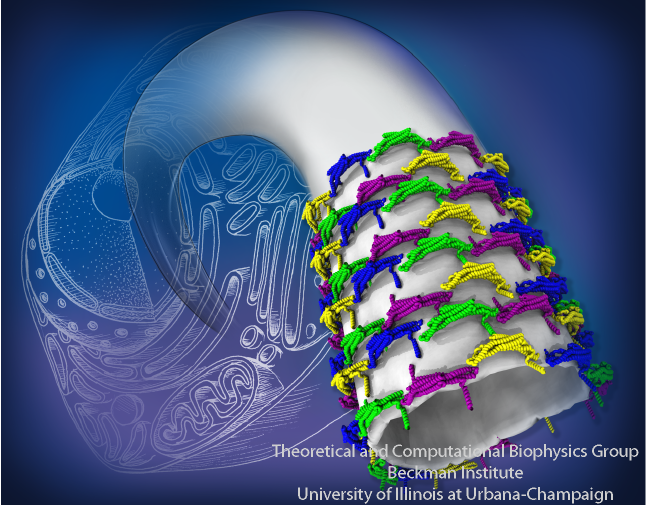
Living cells are characterized by a great diversity of separate internal spaces, the boundaries of which are made of membranes forming convoluted surfaces of manifold shapes. Sculpting these shapes is achieved in many cases by proteins. A single protein is too small to bend the membrane into useful shapes, such as spheres or tubes, that measure 10-100 nm, or more, in diameter. Indeed, the proteins work in teams, but exactly how remained a mystery. Now a computational study elucidates the membrane-sculpting process for proteins called amphiphysin N-BAR domain. Simulations performed with NAMD had revealed a first glimpse earlier (see the Sep 2008 highlight). The new study showed that multiple N-BAR domains form lattices maintained through electrostatic interactions. Positively charged, banana-shaped surfaces of individual proteins bend the negatively charged membrane, while the lattice formation ensures a uniform bending force across a wide membrane surface. In a dramatic example of computational "microscopy" the 200 microsecond sculpting of a large flat membrane into a complete tube was observed. More here.
Computational modeling seeks to simulate biomolecules, particularly proteins. The dream of computational biologists is that their simulations realistically mirror the structure and dynamics of proteins, which act as molecular machines in living cells. Indeed, when researchers tried to simulate how a nascent protein folds into its known shape the chosen protein, called WW domain, did not fold properly (see the May 2008 Highlight). Until recently, simulations could follow protein movement for about 0.0000001 seconds and the computational mirror seemed to work well. Recently, however, simulations with the program NAMD began to follow proteins for almost 0.0001 seconds, a thousand times longer, and the mirror showed cracks. Thus, a question arose as to what went wrong and how the distortion could be repaired. It was unclear whether the simulations still did not last long enough, or whether the physical interactions in the protein were poorly described in the computer model that was used. As reported in a recent paper, the interactions show subtle errors, significant enough to throw off the energy balance in the folding protein. Fortunately, the results suggest ways to improve the computation of physical interactions to fold proteins more accurately, repairing the cracks in the mirror. More information is available at our protein folding website.
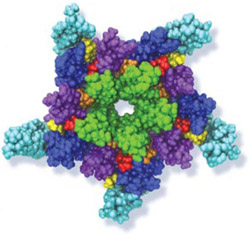
NCSA News Release: Biophysicists at the University of Pennsylvania used NAMD running on NCSA's Abe to clarify a mysterious interaction between cholesterol and neurotransmitter receptors. Research into how anesthesia works may eventually unlock not only that mystery but dozens of others as well. "Anesthetics have improved significantly over the last hundred years, but the mechanism of anesthesia is not understood at all," says Grace Brannigan, a researcher at the University of Pennsylvania's Center for Molecular Modeling (CMM).
To gain insight into how anesthetics work, a team consisting of Brannigan and fellow researcher Jérôme Hénin, University of Pennsylvania professors Michael Klein and Roderic Eckenhoff, and Richard Law of the Lawrence Livermore National Laboratory, is focusing on the nicotinic acetylcholine receptor (nAChR). This receptor, found in both brain and muscle cells, is a ligand-gated ion channel. The channel opens or closes in response to binding with a chemical messenger (ligand) such as a neurotransmitter, like acetylcholine. When the channel is open, ions can cross the membrane. Anesthetics are believed to close the channel, thus reducing sensations and possibly causing the memory loss associated with being under general anesthetic.
The researchers hope their work leads to improved drug design. "You could fine-tune the properties of the drug if you could understand how the mechanism works," says Brannigan. "For instance, by understanding how anesthetics work, you could design new anesthetics that could be more powerful yet maybe wouldn't have some of the side effects that current ones do."
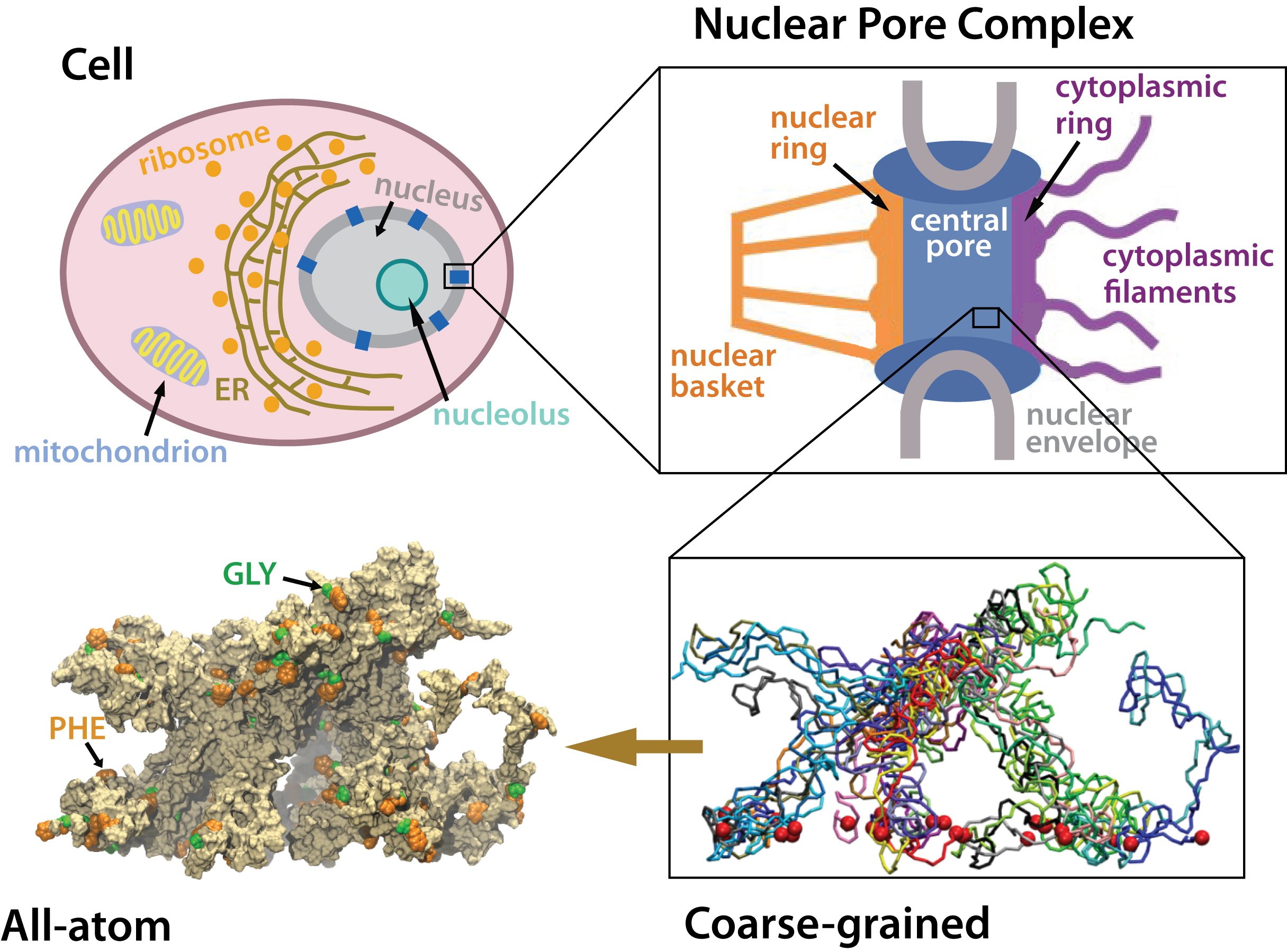
Many living cells, so-called eukaryotic ones, organize their genetic materials in the cell's nucleus, enveloped by a double membrane with guarded access through pores that involve an amazing filter. Like an ordinary filter it permits passage of small particles (biomolecules), but not of large particles (e.g., proteins). However, certain large particles, proteins called transport receptors, can pass. The filter is made of long "finger" proteins anchored inside the pores. The transport receptors can intermittently widen the filter. But to observe how this is achieved is difficult since the finger proteins are highly disordered. As reported recently, simulations using NAMD suggest now a simple and elegant answer: the finger proteins bundle in groups of 2 - 6 and form a brush, filling with its bristles the nuclear pores. The bristles are bundles of finger proteins and have two key properties: (i) on their surface they are dotted with spots of amino acid pairs, phenylalanine and glycine, that are known to interact favorably with transport receptors (see the Aug 2007 highlight, the Feb 2007 highlight, and the Jan 2006 highlight); (ii) the bristles are also interconnected, namely where finger proteins change from one bundle to another bundle, which they do with some frequency. It appears then that the bristles of the nuclear pore filter form an energetically favorable environment for transport receptors. A recent report of new simulations shows that transport receptors are pulled into the bristles of the nuclear pore filter. More information here.
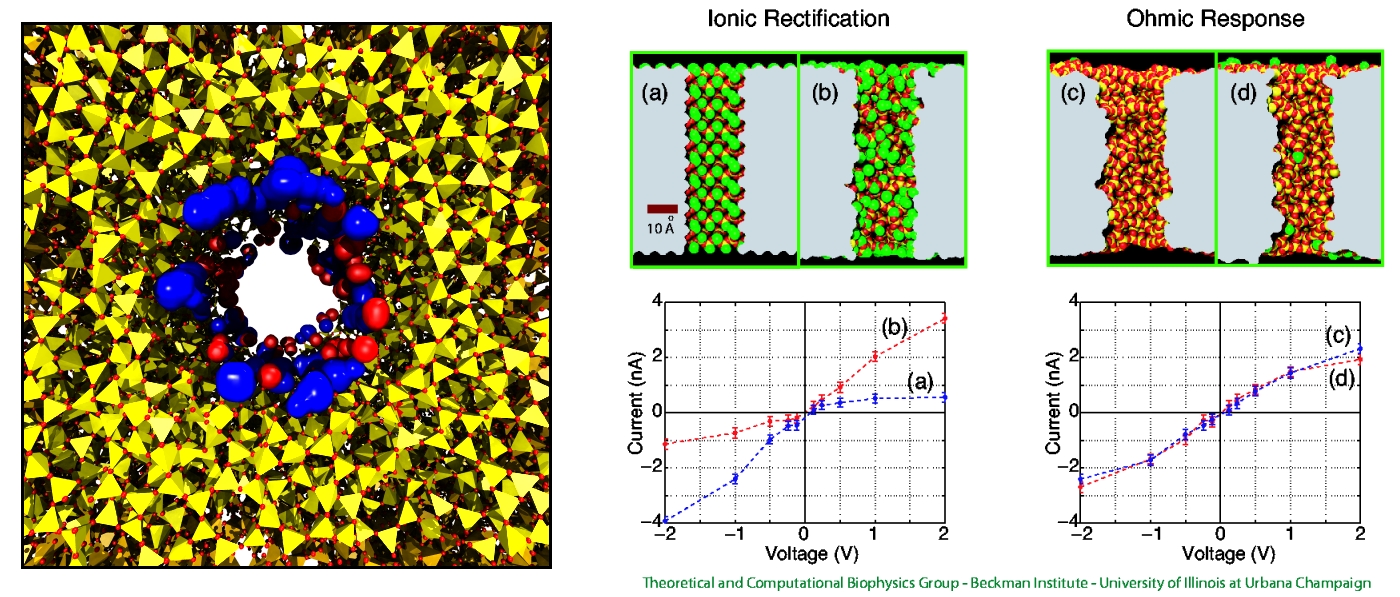
Nanotechnology develops small devices with dimensions below 100 nm, one hundred times smaller than the diameter of a human hair. Nanodevices can be used for a wide range of applications, such as biomedical sensors (see the Jan 2005 highlight) or tools for studying DNA properties (see the Feb 2009 highlight and the Nov 2005 highlight). In building and controlling such small devices, researchers run into problems such as surface effects and significant thermal fluctuations. Furthermore, properties arising from the discrete nature of matter start to dominate at the nanoscale, producing phenomena not observed in larger devices. For instance, when immersed in electrolytic solution and under the influence of an electric field, nanopores act as diodes for ionic currents, conducting in one voltage polarity better than the other, a behavior which has been proposed as the basis for developing nanoelectronic devices. In a recent report, researchers have studied this so-called rectification behavior by means of molecular dynamics simulations using the program NAMD, the ionic rectification inside the nanopore being described in atomic detail. More information can be found here. See also our recent biotechnology review.
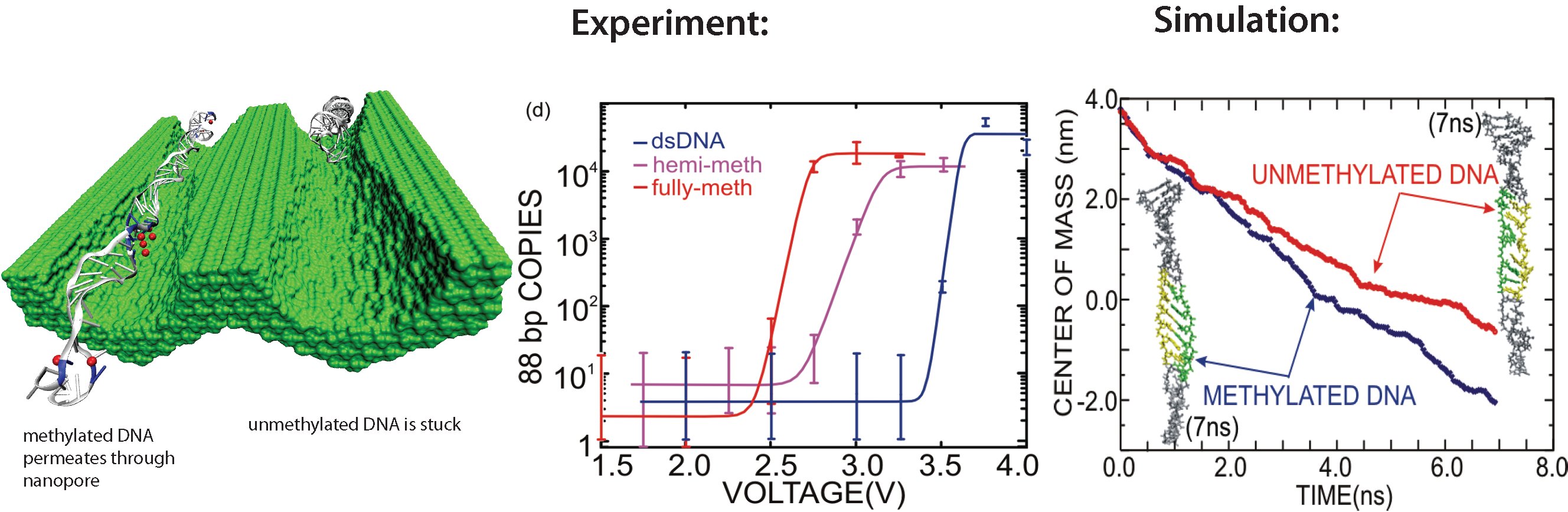
image size:
298.9KB
made with VMD
All cells making up the human body contain the same DNA in their nucleus, the DNA entailing about 30,000 genes and each gene containing instructions for a protein. Despite this sameness, the cells in different parts of our body are very different due to many factors, a key one being that the level of expression of genes into protein is highly regulated and differs strictly from cell to cell. One rather common regulation mechanism involves methylation of one of the four bases of DNA, cytosin. Researchers find that the long DNA in human cells show spots of methylated cytosins, the methylation being correlated with the expression level of the genes near the spots. In fact, medical researcher relate several cancers to improper methylation of DNA. Despite the common occurrence of regulation by methylation, researchers have little understanding how methylation, that changes an H (hydrogen atom) for a CH_3 (methyl group) here and there, i.e., just adds small bumps on a rather bulky DNA molecule, affect the physical properties of DNA such that expression levels are altered. It was found that there are proteins that can recognize the CH_3 groups, i.e., the bumps, on the DNA, but researchers have a hunch that methylation does affect DNA properties directly, i.e., without protein markers, but do not know which properties. In a collaboration between bioengineers measuring the passing of DNA through nanopores and computational biologists simulating this process with NAMD (see also the Nov 2005 highlight stretchable DNA) first hints emerge that methylation does in fact alter DNA's ability to stretch itself through a nanopore. As reported recently, pulling DNA electrostatically through nanopores is easier for methylated than for unmethylated DNA, as seen both in experiment and simulation. The findings promise insight into an important chapter in the field of genetic control. More on our methylated DNA website. See also our recent biotechnology review.
image size: 604.1KB
The ribosome is one of the largest molecular machines present in hundreds to thousands of copies in every cell, in charge of synthesizing every protein in the cell faithfully from genetic instruction. For this purpose the ribosome "reads" the sequence of bases on so-called messenger RNA, three bases at a time and depending on the base triple, the codon, elongates a nascent protein by one of 20 possible amino acids, avoiding to an impressive degree adding a wrong amino acid. So far one knew that the reading is done by transfer RNA molecules that have "foots" which match the possible codons and a "head" that brings along the associated amino acid. Each amino acid has its transfer RNA, the transfer RNAs checking if the next codon is "theirs," and if it is they add the proper amino acid to the nascent protein, elongating it. But how does the ribosome make the critical decision at the decoding center, namely if the transfer RNA "foot," the so-called anticodon, matches the codon? The answer is not known, but a key detail has now been discovered through a combination of electron microscopy and molecular dynamics simulation using NAMD, VMD, and a method called flexible fitting (MDFF, see the June 2008 highlight). It was known that a third molecular system is involved, called the elongation factor Tu (EF-Tu), which generates a key signal to the ribosome and transfer RNA through a chemical reaction. This reaction involves chemically attacking a substrate of EF-Tu, the molecule guanosine-triphosphate (GTP), with water, breaking a bond and turning GTP into guanosine-diphosphate (GDP). The puzzle was that EF-Tu is far away from the decoding center. The collaboration between experiment and simulation, reported here, revealed that "correct recognition" through anticodon-codon binding opens a gate in the EF-Tu that allows water access to the GTP inducing the signaling reaction. The finding promises to now establish how the decision at the decoding center is made and how an "open sesame" order is transmitted to EF-Tu. More on our ribosome website.
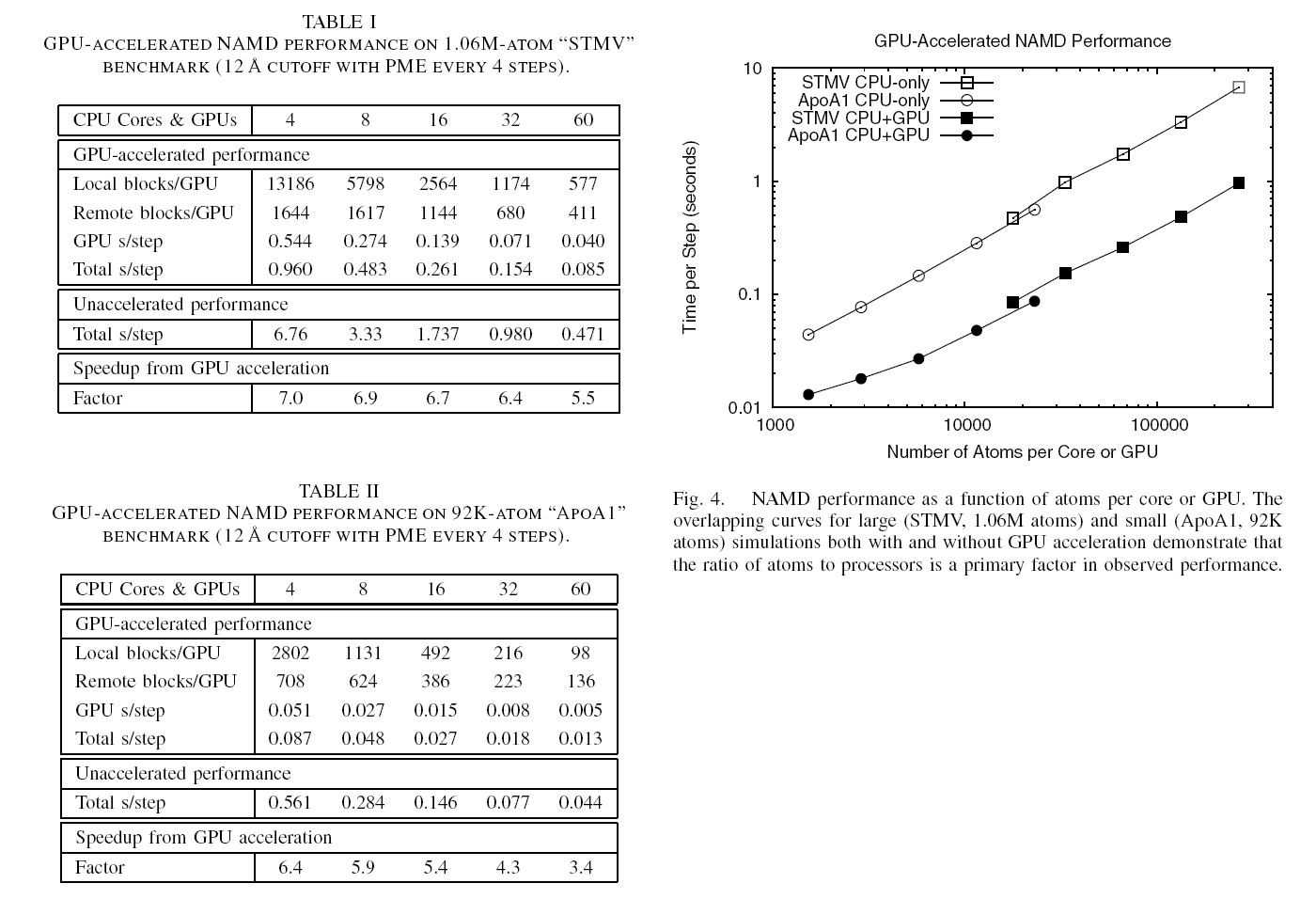
image size: 71.2KB
The most powerful compute engine in your laptop and home computer is what? The central processing unit, of course. Wrong, it is the graphics processing unit (GPU)! Since GPUs are specialized for graphics and games, popular uses of modern computers, their development benefitted from strong market forces. Modern GPUs reach Teraflop speed, passing entire computer clusters filling a room. Unfortunately, GPUs lie dormant when computers are employed for intensive biological computing like biomolecular simulations. GPUs are now getting a wake-up call from biomedical researchers who then enjoy many-fold speed-ups of their laptop computations (see the Oct 2007 highlight), vendors already now hawking GPU-powered "desktop supercomputers." The next challenge is to bring the GPU from the laptop to computer centers, speeding up the world's fastest supercomputers. Building on previous development experience of NAMD and VMD, a recent report describes NAMD running at impressive speed on a GPU cluster. The report suggests techniques, useful also for programmers of other applications to efficiently accelerate computer clusters through GPUs. The report is particularly timely with a new, large GPU-accelerated computer cluster coming on-line. The reported GPU-based speed-ups will permit biomolecular simulations largely unfeasible so far for studies of entire virus particles and cellular organelles. More information here.
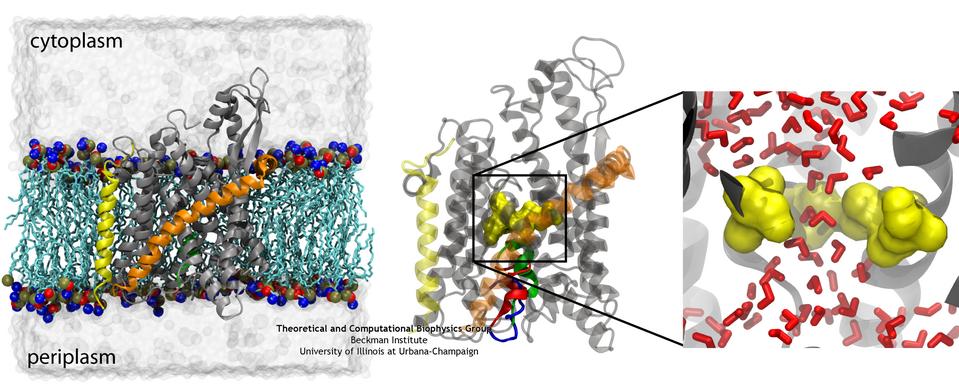
image size:
78.4KB
made with VMD
If you have a leaky roof, you need to plug the hole quickly to avoid further damage. Similarly, cells do not tolerate open holes in their surrounding membranes for long. However, cell membranes do require channels for various molecules to get across, and proteins are no exception. The protein-conducting channel SecY, a membrane-bound protein itself, is the pathway used by other proteins to cross the membrane. When it's not in use, it needs to have a water-tight seal, which is provided by two elements in the channel, a bulky plug and a constrictive pore ring (see the April 2006 highlight). But do these two elements, plug and pore ring, function separately to provide two independent barriers or together, providing a single barrier? Molecular dynamics simulations on the channel and two mutants in which a portion of the plug was deleted have provided the answer, as reported in a recent publication: both components are necessary to prevent leaks in the channel. More information on the amazing channel can be found on our Protein Translocation website.
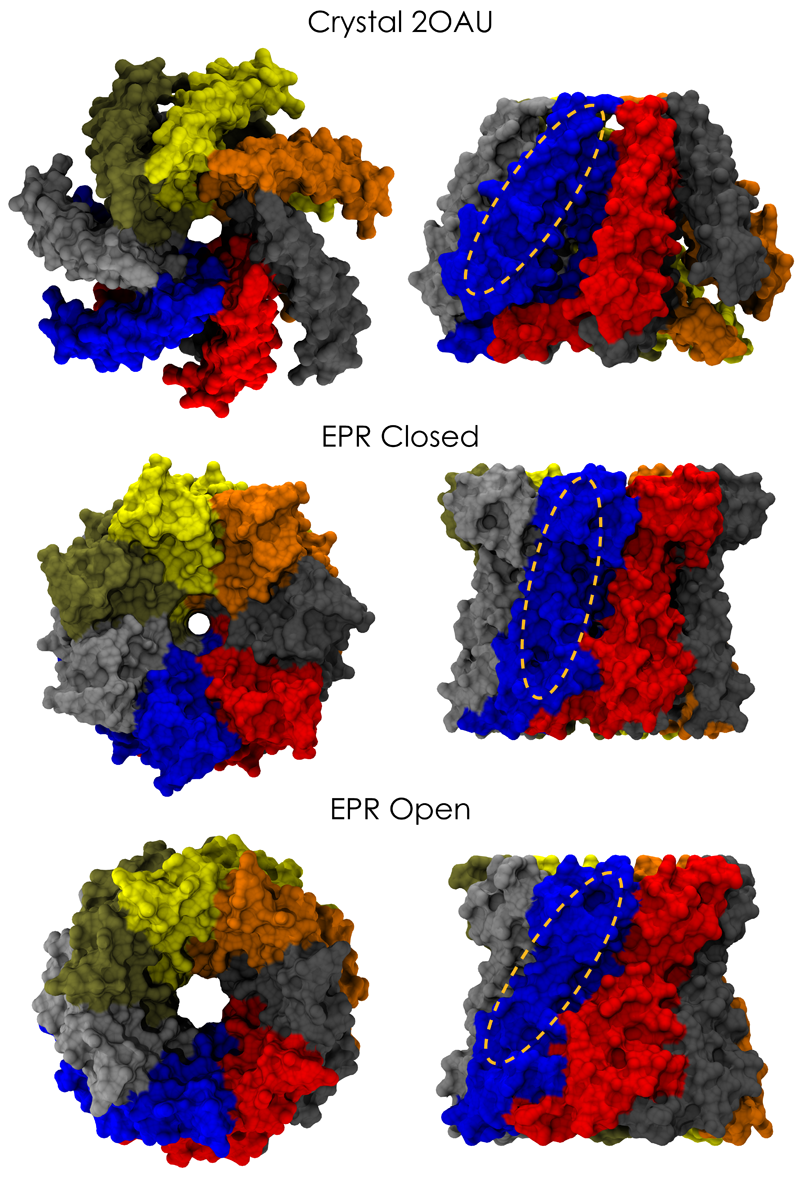
image size:
661.9KB
made with VMD
Biological cells are surrounded by a highly versatile, yet very feeble cellular membrane and need to balance differences between the cell's interior and exterior that otherwise would burst the membrane. For example, the osmotic pressures inside and outside the cell need to be closely balanced. Thousands of proteins in the membrane act as gatekeepers, opening pores that can also act as safety valves, helping to reduce the interior-exterior difference in pressure rapidly. One such protein, the mechanosensitive channel of small conductance MscS (see the Mar 2008 highlight, "Observation and Simulation Depict Cell's Safety Valve", the Feb 2007 highlight, "Observing and Modeling a Crucial Membrane Channel", the May 2006 highlight, "Electrical Safety Valve", and the Nov 2004 highlight, "Japanese Lantern Protein") opens in response to cellular membrane tension generated due to a drastic imbalance in osmotic pressure as it arises when a bacterial cell suddenly finds itself in fresh water, rather than a highly saline physiological medium. The MscS channel widens then to jettison molecules out of the cell and reduce tension on the cellular membrane quickly. In a recent report, researchers have combined experimental data from electron paramagnetic measurements and computer modeling to reveal in atomic detail how MscS opens and closes its channel. Combining measurement and modeling, the researchers established a highly resolving computational microscope, unmatched by existing microscopes (more on our MscS website).
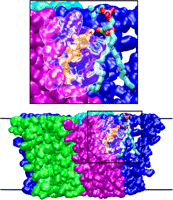
University of Pennsylvania News Release: Biophysicists at the University of Pennsylvania have used 3,200 computer processors and long-established data on cholesterol’s role in the function of proteins to clarify the mysterious interaction between cholesterol and neurotransmitter receptors. The results provide a new model of behavior for the nicotinic acetylcholine receptor, a well studied protein involved in inflammation, Alzheimer's disease, Parkinson's disease, schizophrenia, epilepsy, the effect of general anesthetics and addiction to alcohol, nicotine and cocaine.
Moreover, the results apply to closely related receptors that bind serotonin and GABA, which are neurotransmitters directly involved in regulation of mood and sleep.
The findings have broad implications for, among other fields, pharmacology. Drug development in this arena has to take into account the structure and chemical makeup of this receptor, both of which researchers now say were incomplete. Drugs acting on the receptor have been thought to interact with the protein as though it were isolated.
Now, researchers believe that drugs binding to the receptor not only interact with amino acids — the building blocks of the protein receptor — but also cholesterol tucked away within the protein. The shift in thinking transforms the understanding of this receptor in many ways, from shape and structure to its interaction with its environment and its response to neurotransmitters. The new model should spark a reexamination of several decades of research on the receptor's structure and function.
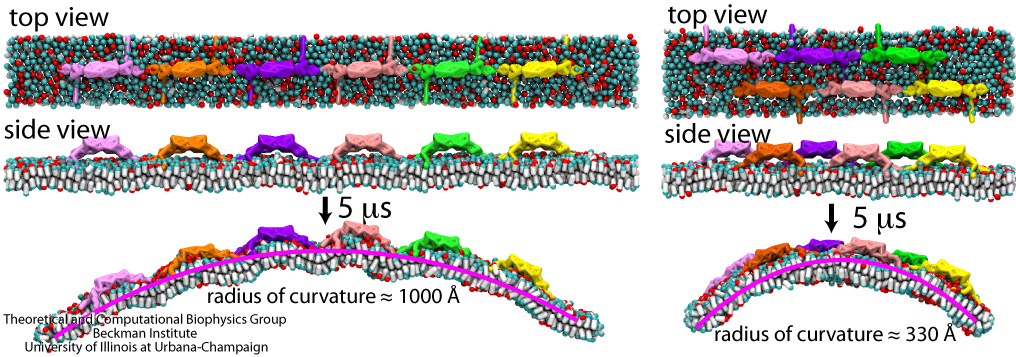
image size:
287.1KB
made with VMD
Living cells organize many functions: molecular import and export, signaling, transcription of genes into proteins, movement, building, repair, and more. These functions are realized through a complex architecture of the cell interior reflected in a system of labyrinthine membranes forming manifold cellular organelles from tubes, vesicles and many other shapes. Accordingly, cells need to sculpt their membrane in never ceasing processes and have at their disposal a wide range of mechanisms. A key sculpting mechanism is furnished by proteins, so-called BAR domains, that apparently form lattice-like scaffolds adhering to membrane surfaces. Such scaffolds have been observed through electron microscopy and they are now also being described through molecular dynamics simulations using NAMD. As recently reported, the simulations, carried out at four different levels of resolution (from an atomic to a continuum level), revealed that different arrangements of BAR domains lead to different curvatures. The simulations help to explain why BAR domains working in teams, i.e., in lattice formation, sculpt intra-cellular membranes into different shapes, depending on the exact arrangement. An arrangement of BAR domains that is particularly efficient in bending membranes was identified. More information can be found on our BAR domain web page.
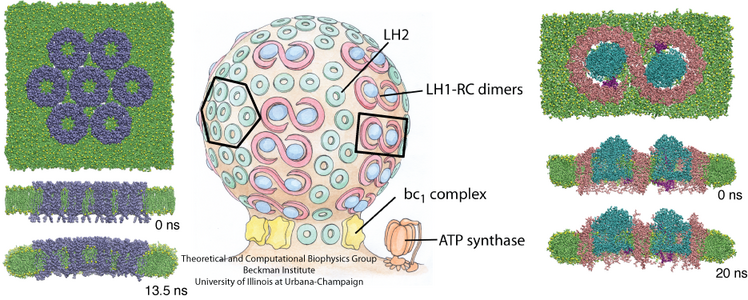
image size:
360.9KB
made with VMD
Chromatophores are the photosynthetic machineries of bacteria. Each chromatophore contains, embedded in a membrane, all the photosynthetic proteins needed to absorb sunlight and turn it into chemical fuel. Chromatophores come in different shapes: while some chromatophores are spherical, others are flat or tubular. It has puzzled scientists how all these different geometries arise, and a hypothesis has developed that it is the photosynthetic proteins that render the shape of chromatophore membrane. In a study reported recently, computational biologists using NAMD took an atomistic look at how the chromatophore proteins bend the membrane. Simulations showed that the most numerous photosynthetic proteins dome the membrane, building arched membrane patches that can then be assembled into a spherical chromatophore. These simulations demonstrated that photosynthetic proteins construct their individual membrane environment, and when many of such proteins come together in the bacterial membrane, they can build functional cellular units with unique geometries. For more details, please see our chromatophore website.
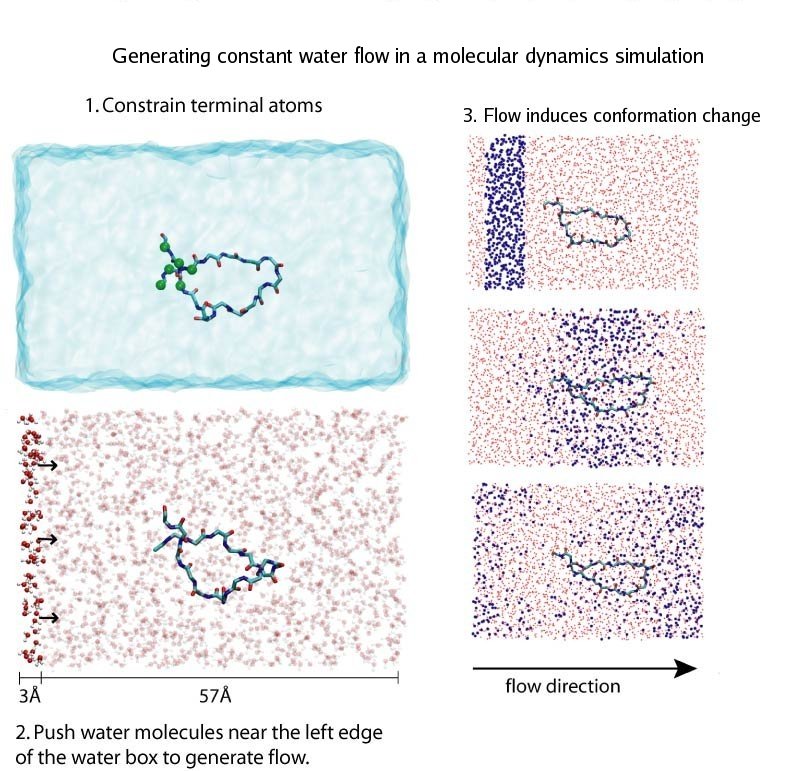
Ouch!! You cut your finger with a knife. Blood immediately starts coming out from the wound, and in a panic you search through your drawer for a band-aid. However, well before you can find the band-aid, and even before the "ouch" came out, just about the same time you sensed the pain, your body's self-healing mechanism was turned on. A multistep signaling cascade involving a dozen different proteins in the blood started immediately after the cut, calling for platelet cells to clog arround the wound and form a plug to stop the bleeding. How does our body sense a wound so fast? A possible answer is that the platelet cells carry on their surface a sensitive molecular flow sensor, a protein called GP1b, which senses erroneous blood flow caused by a cut in the blood vessel. It has been hypothesized that a small segment located on the alpha-subunit of GP1b, called the ß-switch, transforms from a random coil to a ß-hairpin in the presence of shear flow (see movies). The ß-switch, in its ß-hairpin form, is able to bind to a protein called von Willerbrand factor, which will then anchor the platelet to the damage site. To test this hypothesis, researchers resorted to computer simulation using the program NAMD and mimicked blood flow around GP1b. As reported recently, the researchers discovered that a long suspected part of GP1b indeed changes shape under flow conditions, the likely trigger of the body's self-healing system. For more information see our molecular flow sensor web site.
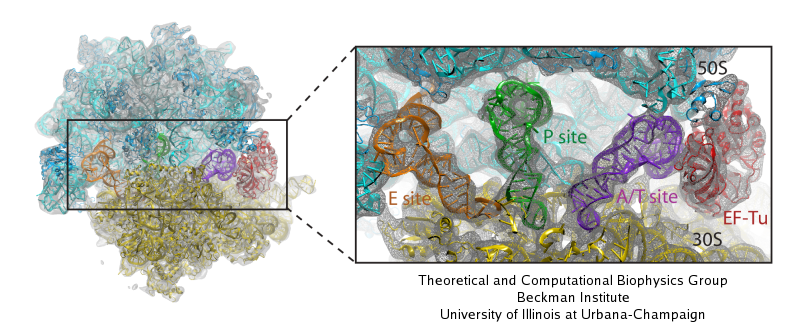
image size:
117.5KB
made with VMD
Living cells are brimming with activity, much of it due to their manifold molecular machines pulling cargo, importing and exporting molecules, digesting food molecules and transforming their energy, reading and copying genetic messages, or synthesizing proteins from these messages (the latter done by the ribosome). Static structures of the molecular machines have been resolved through crystallography: machines pressed into the confinement of crystals and frozen into inactivity reveal their atomic level geometry through this methodology. However, many machines, for example the ribosome, undergo large conformational transitions during their cyclic action, but active motions are hard to view in atomic detail. A way out is offered by electron microscopy which freeze-shocks machines into states characteristic for action cycle intermediates. Unfortunately, the method does not yield atomic resolution images, leaving the chemical detail needed for a comprehension of the mechanisms blurred. Computational methods can be used to bridge the resolution gap: atomic level structures of non-functional states of the machines captured in crystals are deformed to match electron microscopy images. Until recently, the method worked well only for very small machines. Now a team of electron microscopists and computational biologists using NAMD extended the method to common size machines and reported its successful application to the ribosome, providing astonishing detail about ribosome dynamics and function. For more details, see our MDFF website.
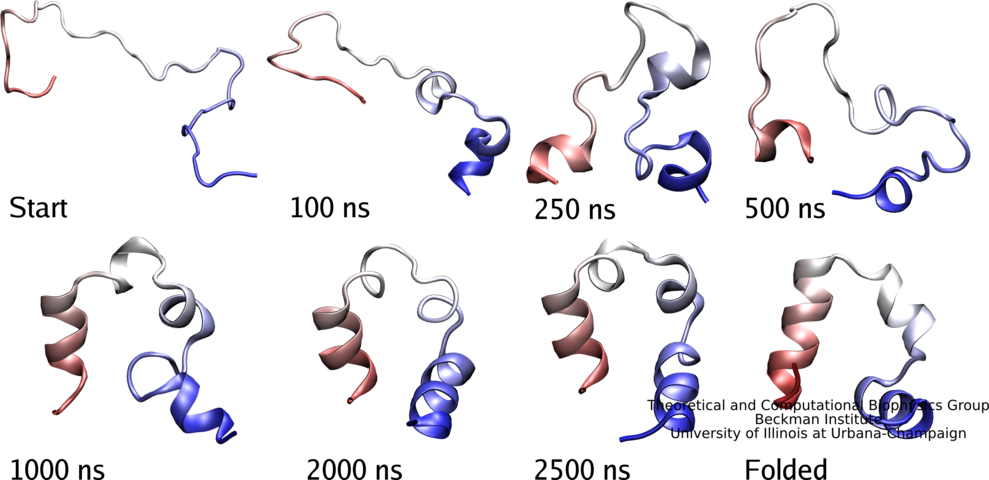
image size:
32.4KB
made with VMD
Proteins carry out most functions in living cells, from import of food substances to chemical synthesis to motion to signaling. Proteins are chains of amino acids like GLSDGEWQLVLNVWGKVEAD... where each letter stands for one of twenty amino acids that are the building blocks of proteins, i.e., G for glycine or L for leucine. In general, sequences of proteins native to cells fold into unique three-dimensional structures capable of executing the proteins' specific function. Living cells store the amino acid sequences of their many different proteins in the form of DNA sequences, safeguarding them in the cells genome. On demand, the DNA sequences are translated according to the famous genetic code into amino acid sequences. The amino acid chains of newly synthesized proteins have to fold into the proper structure, an essential process scrutinized by biologists for decades. The folding process often takes milliseconds or longer, but recently proteins were identified that fold within microseconds. This was still a time too long to be simulated through molecular dynamics which could reveal folding in atomic level detail. However, improvements of NAMD have now made simulations of 10 microseconds possible and in a recent report experimental and computational biologists describe a joint study of a protein segment, known as the WW domain, over this timescale. The great increase in simulation time revealed intricate details of WW domain folding, but also points to a need to further improve the computational model (force field) used to simulate proteins. See also our protein folding web site.
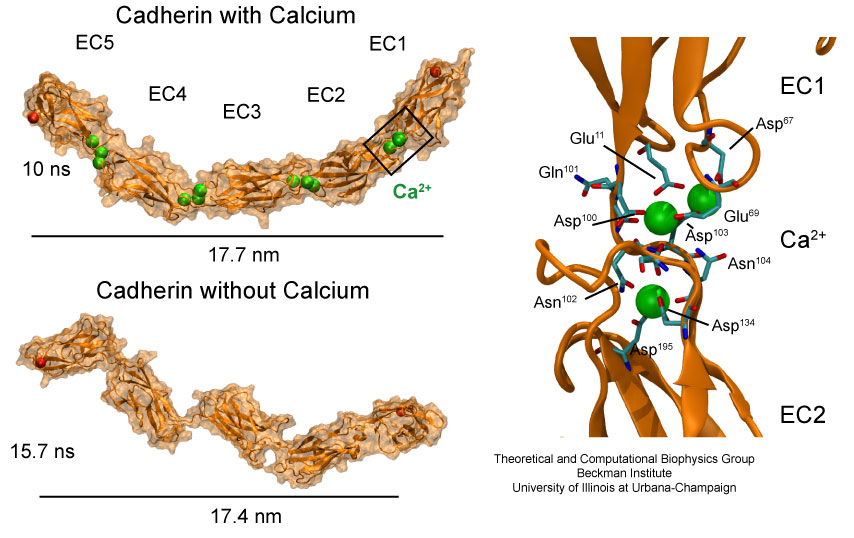
image size:
97.1KB
made with VMD
Adhesion between human cells organizes our body into its organs and parts. The adhesion comes about through an intricate system of molecules that perform their task in a highly selective manner such that the body's different types of cells will find the right cells and stick to them. This selectivity leads to tissue differentiation and the organization of organs as complicated as the brain. Cadherin proteins form a particular family of such adhesion molecules. Interestingly, they glue cells together only in the presence of calcium. Some members of the cadherin family of proteins are also involved in the transduction of sound and cadherin mutants are known to cause hereditary deafness (see the April 2005 highlight, "Hearing: Turning Sound into Voltage"). How cadherins selectively bind to each other and the role of calcium was not well understood, but now molecular dynamics simulations have offered magnificent insight into calcium's role as recently reported. The simulations took advantage of parallel supercomputers and NAMD's ability to harness their power. The simulations revealed that in the absence of calcium cadherins stick out of cell surfaces like ends of loose rope; in the presence of calcium cadherin molecules turn into stiff hooks that link cells together. The calcium-induced links can withstand the strong mechanical forces that arise between cells much larger than cadherin (more on our cadherin website).
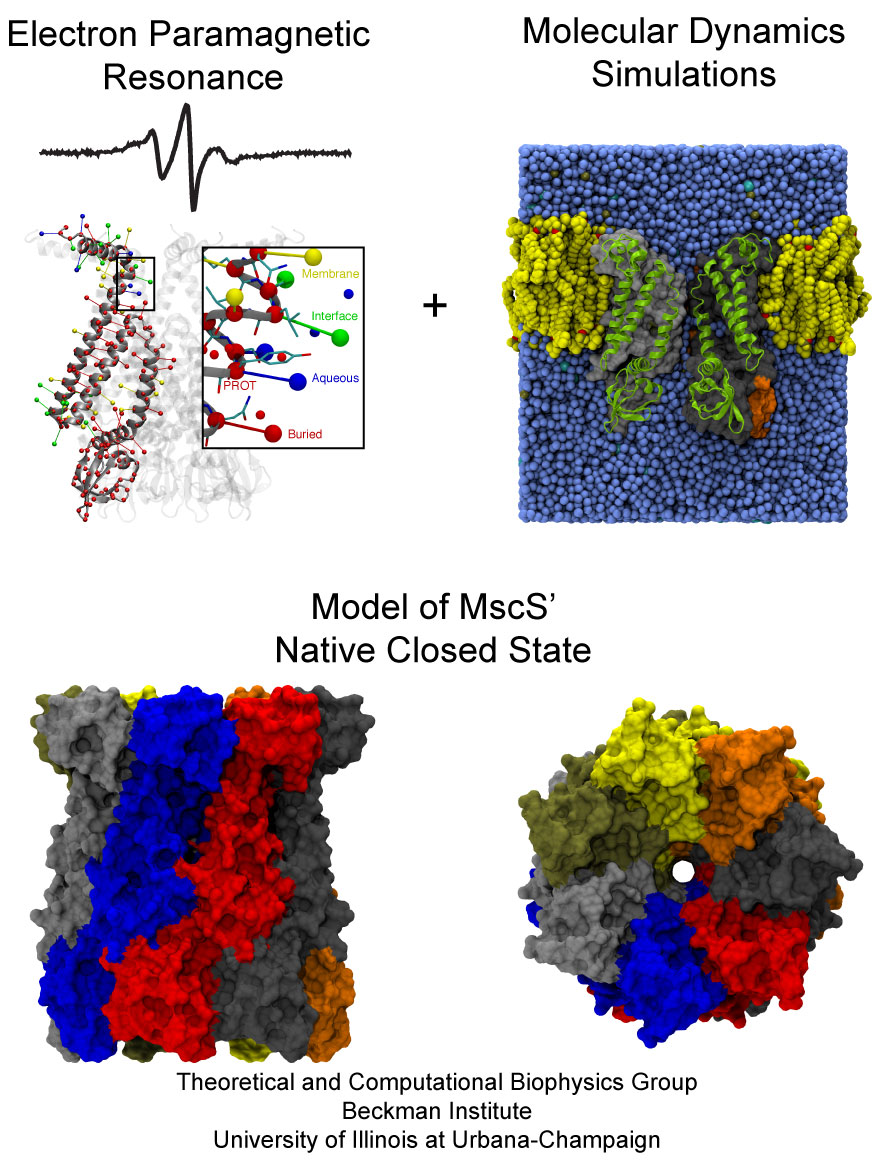
image size:
227.2KB
made with VMD
The environment of cells can undergo drastic changes, for example from dry to wet, in which case cells shrivel or swell. However, they are protected from bursting by a system of safety valves in their cellular membranes that open and release cellular content. Some of the valves open already at low membrane tension, but only little, others open only at higher tension, but wide and without filtering outflow. The mechanosensitive channel of small conductance, MscS, is a low pressure safety valve in bacterial cells (see the Feb 2007 highlight, "Observing and Modeling a Crucial Membrane Channel", the May 2006 highlight, "Electrical Safety Valve", and the Nov 2004 highlight, "Japanese Lantern Protein"). MscS is able to rescue cells about to burst by releasing small solutes through a large and transient opening in the cell membrane, thereby relieving internal pressure. The only way to learn how MscS performs this vital task is by determining its atomic-level structure under native conditions. However, the only structure available for MscS was obtained for the purified and crystallized protein; inspection of the structure left doubt that it shows a functional protein, i.e., a closed safety valve. Now a team of experimentalists and modelers report the structure of MscS seen in its natural membrane environment. In their approach, simulations incorporate information from so-called paramagnetic resonance measurements experiments. This finding is yet another case where the combination of modeling and observation offers entirely new close-up views of living cells (more on our MscS website).
image size:
332.0KB
made with VMD
Bleeding through physical injury is stopped in animals through the formation of blood clots. Such clots, actually arising often in blood vessels without injury, can rupture due to the blood's shear forces and obstruct upstream smaller vessels leading to life threatening stroke, pulmonary embolism, and heart attack. Hence, a blood clot must be both mechanically stable to stop bleeding, yet elastic enough to avoid rupture. Fibrin, the main component of a blood clot, possesses the stated mechanical properties in healthy individuals, but in pathological circumstances needs to be managed through medication. Unfortunately, preventive treatment of blood clots is still a black art since the molecular basis of fibrin elasticity is unknown. Clinicians at the Mayo Clinic teamed up with computational biologists at the University of Illinois to investigate this elasticity, focusing on the protein fibrinogen, the building block of fibrin. The clinical researchers stretched individual fibrinogen molecules measuring the force needed to extend the molecules. They found a characteristic force - extension relationship and its dependence on blood pH and calcium concentration, but they could not interpret the finding chemically, a prerequisite for improving blood clot chemical management. The clinical researchers joined forces with computational biologists who could reproduce the measured force - extension relationship in steered molecular dynamics using NAMD. The simulations starting from known crystallographic structures of fibrinogen offered a full, i.e., atom resolution, chemical picture of fibrinogen elasticity. As reported recently by the clinical and computational researchers the insight gained opens new avenues for blood clot treatment. For example, it was found that pH and calcium concentrations alter the stiffness of blood clots, thereby opening pharmacological avenues for controlling the incidence of pathological blood clots. More on this investigation can be found here.
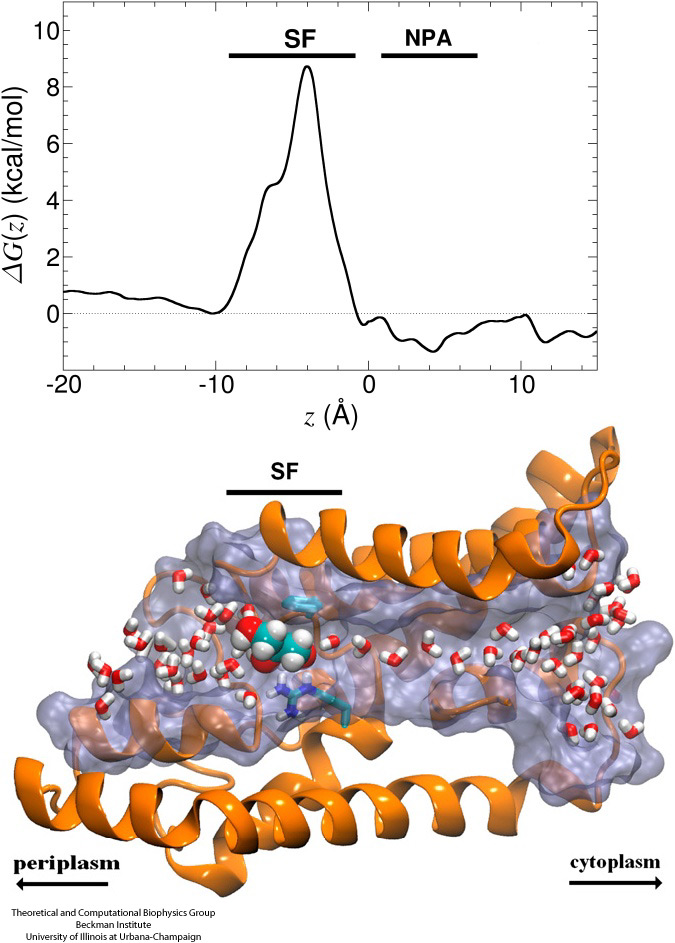
Biological cells protect their interior through their cellular membranes, yet rely on import of nutrients. They have evolved for this import fast conduction channels that include reliable checkpoints distinguishing desirable and undesirable compounds. A checkpoint puts up a veritable obstacle course that only the right compounds can pass quickly. Understanding the channel design is difficult due to lack of detailed experimental data on nutrient dynamics. Presently, the most detailed information comes from viewing channel dynamics computationally, starting from static crystallographic structures. A recent study investigated how glycerols, small nutrient molecules needed by some bacteria, pass through checkpoints realized through the membrane protein aquaporin (see also highlights Gas Molecules Commute into Cell - Mar 2007, Aquaporin and the Cambridge Five - Oct 2006, Cellular Faucets - Feb 2006). Aquaporin furnishes four parallel channels that were monitored computationally using NAMD and a novel algorithm that explores the channel energetics quickly enough to be methodologicaly feasible on today's computers. The results show how the physical characteristics of glycerol, for example the molecule's ability to form so-called hydrogen bonds, its electrical dipole moments, its diffusive mobility and intrinsic flexibility are probed along the channel, discriminating glycerol from other molecules. More on computational investigations of aquaporin here.

Ever since the 1953 discovery of DNA's double helix structure researchers wondered how the double strands are separated so that genetic information stored inside the helix can be delivered from generation to generation. A class of proteins known to achieve this separation are DNA helicases (see Sept 2006 highlight), molecular motors that operate at a fork where a double stranded DNA separates into two single stranded DNAs. Helicases translocate along one of the single stranded DNAs, pushing forcefully into the fork to split the double stranded DNA apart further. Helicases seem to work, though, both through force and through persuasion, exposing to the double stranded DNA a surface that is apparently conducive for strand separation. This property suggests itself on account of the fact that many of the amino acid side groups at the relevant surface are highly conserved among species or evolved from species to species through pairwise mutation. But what chemical strategy evolution had in mind in molding the surface was not realized. Recently, however, researchers seeking artificial means of splitting double stranded DNA apart might have found a key clue. They pulled double stranded DNA at one of its single strands by means of an atomic force microscope from DNA's native salt water environment into a so-called non-polar solvent. The force - distance curve measured suggested that the DNA actually split apart, but there existed no direct experimental means of viewing the splitting. The researchers employed instead molecular dynamics simulations, using NAMD, that indeed clearly revealed the splitting of the DNA strands at a water - oil (octane) interface as reported in a recent publication. The study suggests how helicases achieve the splitting of DNA strands, namely by altering the local environment of DNA from water-like (hydrophilic) to oil-like (hydrophobic). More information here.
image size:
323.1KB
made with VMD
Globins are oxygen-storing proteins, vital to life. In our blood, hemoglobins carry oxygen from our lungs to every cell in our body. In our muscles, myoglobins keep reserves of oxygen to make sure it is available when needed. In some plants, leghemoglobins capture oxygen molecules that would otherwise be harmful to the production of ammonium necessary for the plant's survival. All these globins possess an iron-containig "heme", that grabs on to oxygen for a short time, and share the same protein architecture, despite large variations in their sequences. Since the heme group is buried inside a globin, scientists wondered how oxygen makes its way inside the protein to reach it. Exploring the motion and energetics of globins using the program NAMD researchers learned to gather data that permitted them to visualize, utilizing the VMD software, all the pathways taken by oxygen migrating inside whale myoglobin (see the Aug 2006 highlight and related publication). However, when the researchers turned their attention to the rest of the globin family to compute their oxygen pathways, they found, on their computational spelunking trip, something surprising. Given the conserved architecture of all globins, they expected to see similar oxygen pathways throughout the globin family, but they saw the opposite! Aside from a conserved pocket right at the heme binding site, the distribution of oxygen pathways showed very little similarity from one globin to the next. This result is described in a recent report, which shows that oxygen-pathways are not conserved by evolution, and that their location is not determined by a protein's overall architecture, but rather by its local amino acids. The researchers also learned which amino acids are found more often than others lining oxygen pathways, recognizing that bulky side groups are not hindering, but favoring oxygen passage. More information can be found here.
image size:
221.8KB
made with VMD
Modern computers include a massively parallel graphics processing unit (GPU) designed to perform geometry and lighting calculations at blistering speeds. State of the art GPUs can perform 0.5 teraFLOPS with their hundred cores. The tremendous computational power of GPUs was untapped by scientific computations because it could only be accessed with difficulty until now. As reported in the Journal of Computational Chemistry, recent advances allowing GPUs to be used for general purpose computing have boosted the performance of a number of molecular modeling applications, including NAMD simulations and VMD electrostatic potential calculations. The accelerated versions of these applications run five to one hundred times faster than on the best CPU-based hardware, allowing a single desktop computer equipped with a GPU to provide processing power equivalent to an entire, large computing cluster. More information on GPU acceleration of molecular modeling applications is provided here.
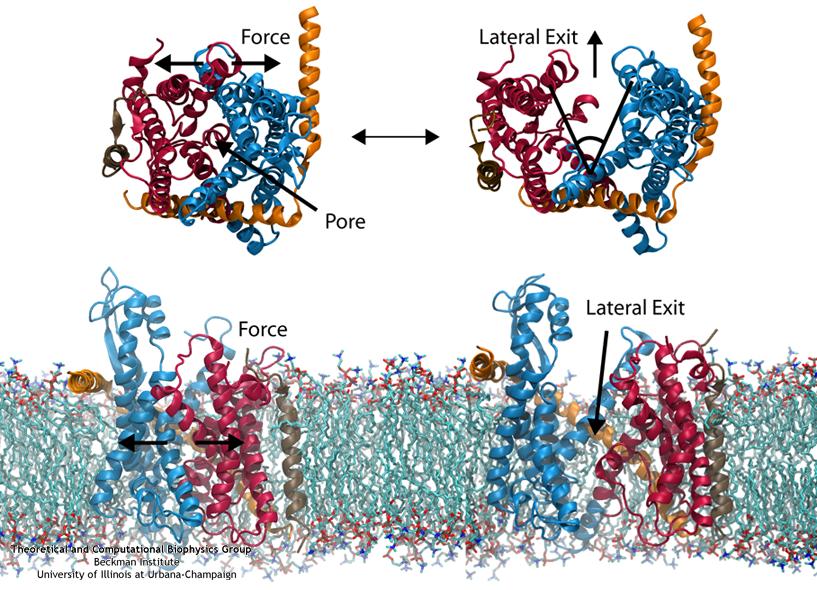
image size:
111.2KB
made with VMD
Everyone knows oil and water don't mix. Proteins observe this rule, too, some choosing to stay in the watery cytoplasm and others choosing the oily membrane. But getting into the membrane is not easy, and most newly formed proteins require another protein, the membrane-bound translocon, to help them insert into the membrane. The translocon, surprisingly also serves as a conduit for proteins across the membrane, thus carrying out a unique dual function. The structure of the translocon showed evidence of a likely "lateral gate", i.e., an exit from the channel into the membrane. How the channel opened to the membrane though, and how it closed afterwards, were not clear from the structure alone. Now, molecular dynamics simulations performed with NAMD, covered in a recent publication, have permitted researchers to understand how the channel opens laterally, how it closes, and how the oily lipids are prevented from invading the water-filled pore. Furthermore, the novel simulation technique, residue-based coarse graining, allowed the researchers to simulate the lipid-channel interactions for up to one microsecond, clearly illustrating that the lipids do not want to mix with the channel interior. More information on these results can be found on the Protein Translocation website.
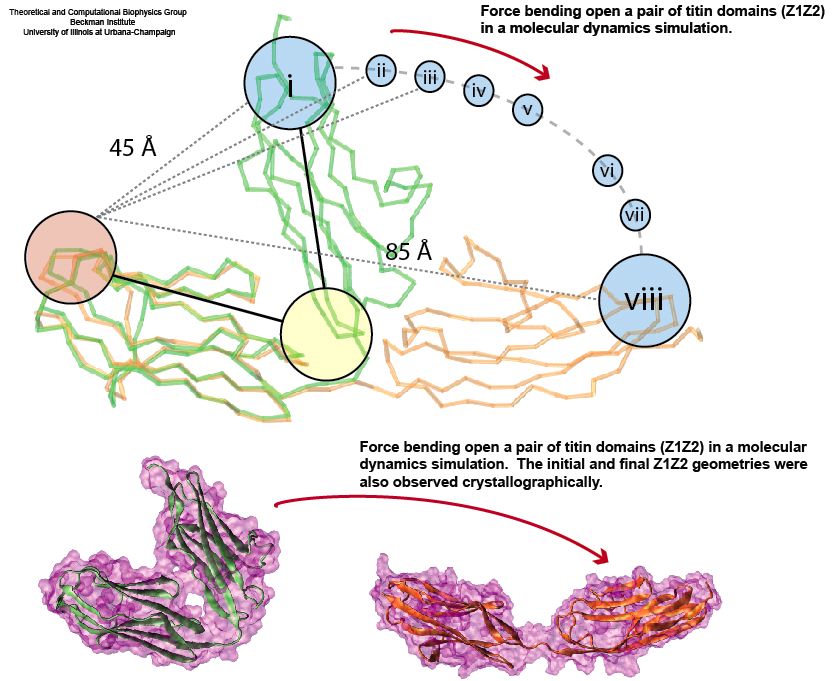
image size:
92.2KB
made with VMD
Muscle fibers, in contracting and extending, generate tremendous force that needs to be buffered to protect muscle from damage. This role falls to the protein titin, with about 27,000 amino acids the longest protein in human cells. Titin functions as a molecular rubber band, but unlike uniform rubber bands, titin is made from over 300 different protein domains strung into a chain. While experiments have found that the individual domains of titin feature remarkable resilience against mechanical stretching, little is known about the elasticity of the overall titin chain. Crystallographers teamed up with computational biologists to investigate this elasticity, focusing on two adjacent titin domains. Molecular dynamics simulations using NAMD suggest, as reported recently, that the overall elasticity of the titin chain stems in part from a zigzag, i.e., accordion-like, motion: as titin is contracted and extended, energy is stored and released in the angular tilt of adjacent domains. More on this investigation can be found here.
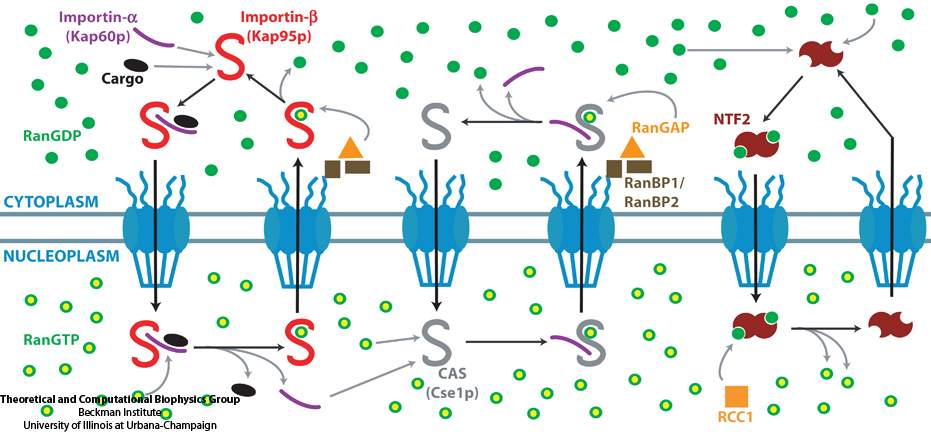
The cells of higher organisms store their genetic material, the genome, in the so-called nucleus where they organize transcription of DNA into messenger-RNA, the blueprint for proteins. The messenger-RNA leaves the cell to be decoded by ribosomes that synthesize the respective proteins. Transcription factors, also proteins, control in the nucleus which parts of the cells' genomes are transcribed. Naturally, the access to the nucleus as well as exit from it must be restricted to transcription factors and related biomolecules. This is achieved by the nuclear pores, wide channels lined with brushes of polymers. The polymers are disordered proteins and prevent passage for most cellular proteins, except for so-called transport factors which bus transcription factors, messenger RNA, and certain larger biomolecule into and out of the nucleus. How transport factors are permitted to pass the nuclear pores, despite many studies, has been largely unknown. Molecular dynamics simulations, based on relevant crystallographic structures, using NAMD provided a comprehensive picture on the passage mechanism as reported recently. The simulations, analyzed with VMD, revealed that transport factors are dotted rather regularly on their surface with spots that bind to the brushes of nuclear pore proteins. While any protein may accidentally exhibit such a binding spot or two, only transport factors offer a regular pattern of such spots on their surface that apparently is their passport permitting them movement into and out of the nucleus, i.e., helping them to glide through the pores' protein brushes. More on simulations of transport factors can be found here.
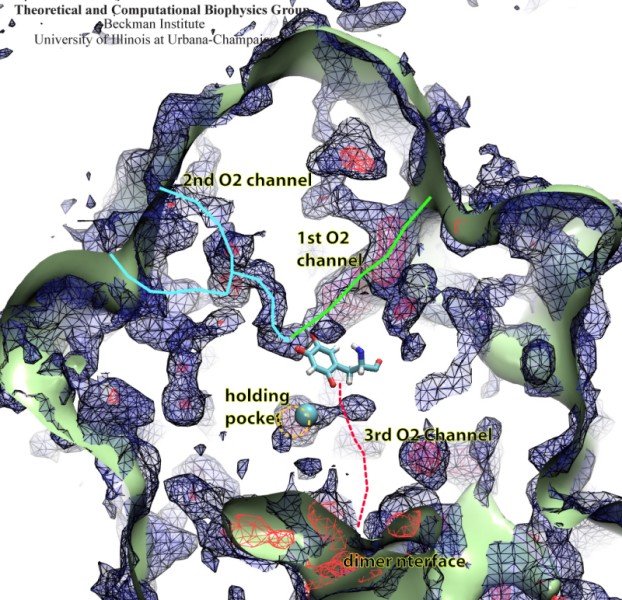
image size:
133.1KB
made with VMD
Because oxygen gas is very reactive, it is frequently employed by the cell as a reagent by proteins called enzymes, which build the organic compounds that the cell needs. One such enzyme belongs to the copper amine oxidase family. These proteins transform amine-containing compounds into molecules needed by the cell, by reacting the compounds with oxygen. Researchers have long been interested in finding out how the various reagents reach the buried copper active site before the final oxidation reaction can occur. While copper amine oxidases exhibit an obvious channel for capturing the amine compounds to be modified, it had been unclear until now how oxygen molecules make their way through the enzyme. With the help of computer simulations using NAMD, researchers have identified in a recent publication, the routes taken by oxygen inside various copper amine oxidases from different species. In order to accomplish this, they analyzed simulations of the motions of four copper amine oxidases, using the VMD analysis and visualization software, which can predict the probability of finding oxygen molecules anywhere inside the simulated proteins. This analysis found numerous oxygen conduction routes inside each copper amine oxidase, i.e., oxygen can enter the protein through many routes, as it would in a sponge. More information on finding O2 migration pathways in proteins can be found here.
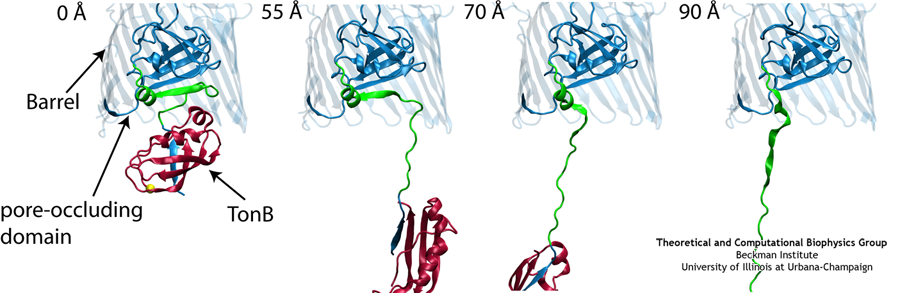
Like all organisms, bacteria have to eat. However, bringing nutrients in from the outside world is not an easy task for many bacteria that are surrounded by an extra membrane. The second membrane, called the outer membrane, offers additional protection but at a cost: no energy can be generated or stored at the outer fringes of the cell. So, to import large, rare nutrients that cannot cross by diffusion alone, bacteria have evolved a unique transport system which couples the inner, energy-generating membrane to the passive outer membrane, known as the TonB-dependent transport system. TonB, an inner membrane-associated protein, transfers energy across the periplasm to a variety of outer-membrane transporters. These transporters have a large, beta-barrel structure which is blocked in the middle by a plug called the 'luminal domain'. How TonB transfers energy to the transporter and causes the luminal domain to come out is still a mystery though. Now with the help of computer simulations using NAMD and a recent crystal structure of TonB coupled to BtuB, the transporter responsible for vitamin B12 transport, researchers have shown that TonB can mechanically activate the transporter by pulling on the luminal domain, causing it to leave the barrel. Using steered molecular dynamics, it was found that TonB stayed firmly attached to the luminal domain of BtuB, even though the contact between the two is limited to just a handful of residues. Furthermore, this pulling initiated unfolding of the luminal domain, opening a transport pathway for the substrate. These results, the subject of a recent publication and also highlighted in Science, demonstrate how a mechanical coupling can bridge the gap between the two membranes, thus enabling outer membrane transport.
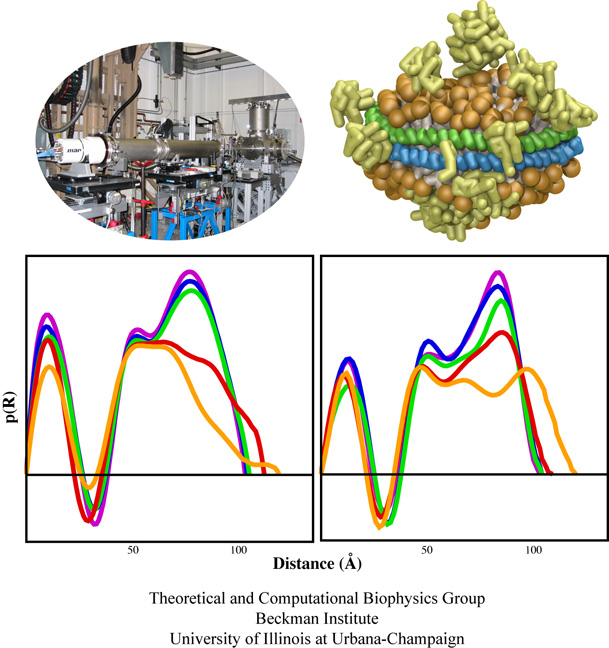
We all know sushi rolls, but just to be sure here is an easy definition: a wrapper encircles rice which holds a precious bit of fish. To make a sushi role is an art and the same holds true for molecular sushi that is made of two lipoproteins as wrapper, lipids as rice, and membrane proteins as filling. Sushi rolls are for eating. Molecular sushi roles are for holding membrane proteins in place for physical analysis; they actually come only in sliced form, one disc at a time. Due to their size, the discs are called nanodiscs. Since membrane proteins are notoriously difficult to study experimentally due to their need to be in a "native" membrane environment, nanodiscs are a great tool, furnishing a membrane environment that has been used to embed a variety of membrane proteins for biochemical assay, including cytochrome P450's, rhodopsin, bacterial chemoreceptors, blood clotting factors, and translocation proteins. Unfortunately, it is difficult to make either real or molecular sushi rolls (nanodiscs). In either case one needs to lay down the ingredients first. In the case of nanodiscs, one starts from the raw ingredients which are solubilized by the detergent cholate. Removing the detergent allows the nanodiscs to self-assemble. However, the assembly process is difficult to quantify experimentally, thus researchers rather studied the disassembly process, i.e., how detergent disassembles preformed nanodiscs. One can watch a sushi chef make rolls, but watching the disassembly and assembly of nanodiscs is harder due the the small size. Fortunately, a computer can image the process. In a recent publication, nanodisc disassembly through the addition of increasing concentrations of cholate was monitored through computer simulations using NAMD and verified through experimental small-angle X-ray scattering. The study showed how cholate molecules insert themselves at the interface between the lipids and lipoproteins towards complete disassembly. The simulations employed a new method called residue-based coarse-graining. For more information, see our webpage on nanodiscs.
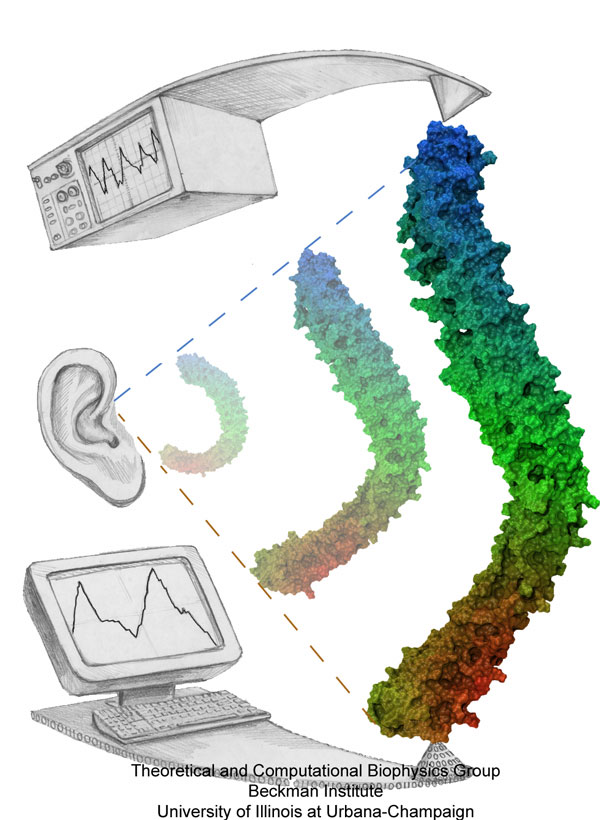
image size:
91.7KB
movie (
789.9KB
)
Mechanical forces are everywhere in human life. Strong forces power machines and cars, our body's forces let us labor and move, soft forces are sensed through touch, even softer ones through hearing. Forces are also ubiquitous in the living cell, driving its molecular machines and motors as well as signaling ongoing action in its surroundings. Man made, force bearing machines rely on extremely strong materials not found in the cell. How can the cell bear substantial forces? Also, how do cells sense extremely weak forces as in hearing, surpassing most microphones? Single molecule measurements, reviewed in a recent issue of Science, begin to answer these questions offering information on biomolecules' mechanical responses and action. However, the information offered by these measurements is not enough to relate the biomolecular function to the biomolecular architecture. Biomolecules in cells can move in amazing ways, but we did not know why. As one contribution in the Science issue demonstrates, computational modeling comes to the rescue. It can simulate the measurements and, in doing so, can reveal the physical mechanisms underlying cellular mechanics at the atomic level. In as far as observed data are available, the simulations show impressive agreement with actual measurements. While initially only following experiments or, at best, guiding experiments, modeling has advanced now further and through simulated measurements discovered on its own entirely novel mechanical properties that were later verified by experimental measurements. Experimentalists reacted to the new competition and began to do simulations themselves. More here.
PSC News Release: University of Utah chemist Gregory Voth and grad student Phil Blood are using PSC’s Cray XT3 to tackle a basic question of endocytosis—the life-sustaining process by which cells absorb material from outside the cell by bending their membrane to form a “vesicle” and engulf it. All animal cells depend on endocytosis, which involves various steps, but begins with curvature of the membrane.
BAR domains are a family of banana-shaped proteins shown to bind to cellular membrane as it curves. Experiments suggest that BAR domains mold their concave surface to a section of membrane and induce a corresponding curvature. Voth and Blood undertook molecular dynamics simulations to look more closely. With the XT3 they’ve been able to run efficiently, using software called NAMD, with as many as 1,024 processors. “The XT3 has been amazing,” says Blood. “We haven’t found a hard limit on scaling up the number of processors.”
They used TeraGrid systems at SDSC, NCSA and University of Chicago/Argonne to construct a model and to explore how long a stretch of membrane they needed for curvature to occur. Their final simulations used the XT3 to include the protein with a 50-nanometer length of membrane—probably the longest patch of membrane ever simulated—for a total of 738,000 atoms. Their results, reported in Proceedings of the National Academy of Sciences (2006), show that the orientation of the BAR domain as it attaches to the membrane determines the degree of curvature.

SDSC News Release: SDSC and UC San Diego researchers are using NAMD to zero in on the causes of Parkinson's disease, Alzheimer's disease, rheumatoid arthritis and other diseases. The April 2007 FEBS Journal cover story offers—for the first time—a model for the complex process of aggregation of a protein known as alpha-synuclein, which in turn leads to harmful ring-like or pore-like structures in human membranes, the kind of damage found in Parkinson's and Alzheimer's patients. The researchers also found that the destructive properties of alpha-synuclein can be blocked by beta-synuclein—a finding that could lead to treatments for many debilitating diseases.
Lead author Igor Tsigelny, SDSC researcher and project scientist in chemistry and biochemistry at UCSD, said that the team's research helped confirm what researchers had suspected. “The present study—using molecular modeling and molecular dynamics simulations in combination with biochemical and ultrastructural analysis—shows that alpha-synuclein can lead to the formation of pore-like structures on membranes.” In contrast, he said, “beta-synuclein appears to block the propagation of alpha-synucleins into harmful structures.”
“This is one of the first studies to use supercomputers to model how alpha-synuclein complexes damage the cells, and how that could be blocked,” said Eliezer Masliah, professor of neurosciences and pathology at UC San Diego. “We believe that these ring- or pore-like structures might be deleterious to the cells, and we have a unique opportunity to better understand how alpha-synuclein is involved in the pathogenesis of Parkinson's disease, and how to reverse this process.”
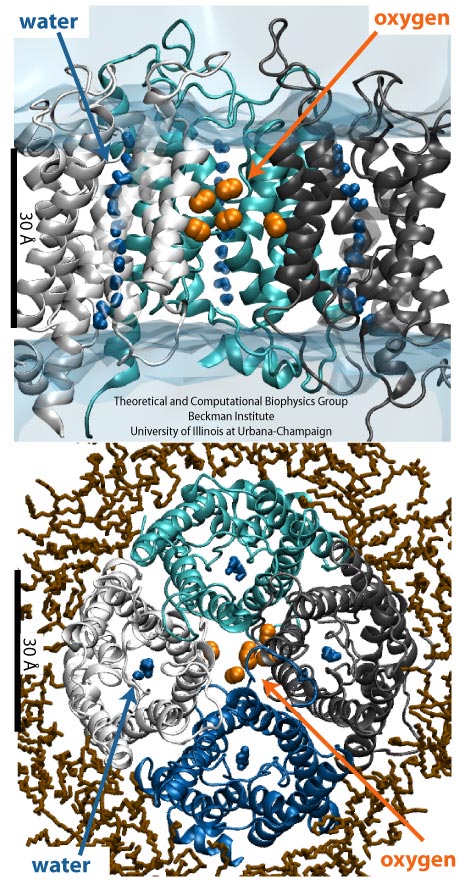
image size:
192.3KB
made with VMD
Every morning, many people drive to work, while others may bike, take the bus or the metro. Similarly, various biomolecules in the human body also reach their destinations in diverse manners. For example, to cross the cellular membrane, small hydrophobic gas molecules diffuse through the lipid bilayer, while water molecules pass through specialized channel proteins named aquaporins (AQPs). Interestingly, just like one may get to work both by bus and by driving, it has been found recently that some gas molecules may have more than one way to cross the membrane, i.e., besides diffusion through lipids, oxygen and carbon dioxide may also pass through AQPs. However, the pathways that these gas molecules take remained elusive. Using molecular dynamics performed with NAMD, researchers have investigated the gas permeability of AQP1 in a recent study with two complementary methods (explicit gas diffusion simulation and implicit ligand sampling). The simulation results suggest that while the four monomeric pores of AQP1 function as water channels, the central pore of AQP1 may serve as a pathway for gas molecules to cross the membrane. More information on the simulations can be found on the aquaporin web page.
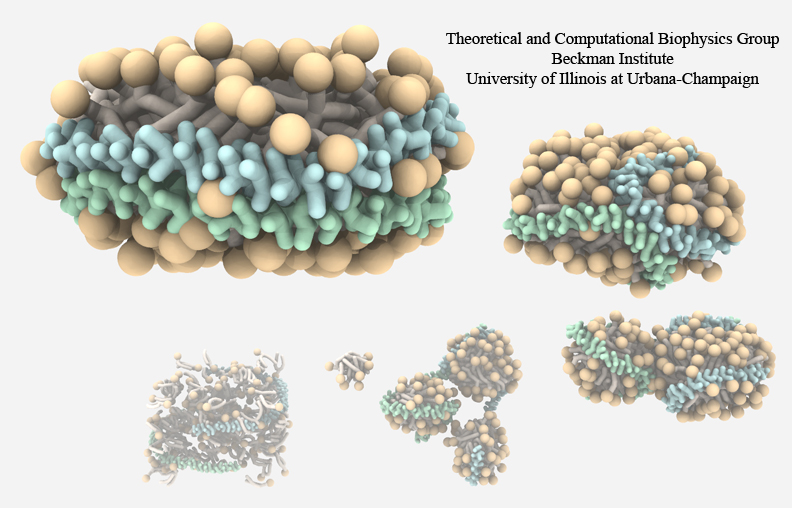
High-density lipoproteins, otherwise known as the "good cholesterol", are the body's way of naturally removing cholesterol in the blood stream. Since lipid and cholesterol molecules are not soluble in blood, lipoproteins are needed to collect and transport them. The proteins wrap themselves around the hydrophobic portions of lipids and cholesterol, effectively shielding them from the aqueous environment and allowing them to be transported through the bloodstream to the liver for degradation. High-density lipoproteins exhibit a variety of shapes and sizes and presently cannot be imaged through experimental observations. Computational methods, however, can provide detailed images of high-density lipoprotein particles, even showing how these particles form in the body. As recently reported (article 1, article 2), so-called coarse-grained molecular dynamics simulations using NAMD discovered how lipid molecules are corralled by lipoproteins to form disc-like high-density lipoprotein particles. The simulations show in remarkable detail the aggregation of proteins and lipids, starting from a random arrangement of lipids that are mopped-up by two lipoproteins, eventually forcing the lipids into a disc-shape surrounded on its circumference by belt-like lipoproteins. For more information, see our webpages on HDL & nanodiscs and coarse-grained modeling.
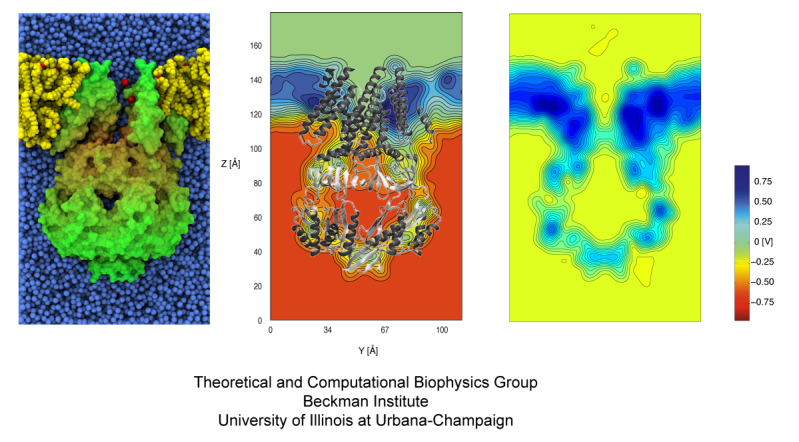
Bacteria employ membrane proteins as crucial safety valves that release water and small solutes under challenging osmotic conditions (see the May 2006 highlight, "Electrical Safety Valve" and the Nov 2004 highlight, "Japanese Lantern Protein"). There are valves for balancing small pressure differences between the inside and outside of bacterial cells, that open and close readily, but there are also ones for protection against large pressure differences as a safety measure of last resort. The valves for balancing small pressure differences, like the one shown in the figure, include a filter that presumably keeps the most valuable molecules inside the cell interior, though this is not understood yet in detail. To reveal the function of such channels a combination of X-ray crystallography, physiological measurements, and molecular dynamics simulations using NAMD has been employed. Crystallography, in a prior study, captured the channel in a half-way open state. Now a team of physiologists and modelers reported the details on valve opening and closing. The experiments, using a pipette small enough to measure currents from a single channel, MscS, along with the simulations revealed that the channel conducts both positive and negative ions when subjected to tension and voltage. The unprecedented comparison of experimental and computational results open a new era of quantitative cell biology that borrows successful research strategies from physics (more on our MscS website).
image sizes:
419.0KB
&
83.2KB
made with VMD
The nucleus is responsible for storing the genome of eukaryotic cells, isolating it from the cellular cytoplasm. Partitioning the genetic material is very important in protecting it from cellular processes or foreign molecules. However, the nucleus also needs to provide access for the rest of the cell to the information stored in the genome. Numerous nuclear pores in the nuclear envelope offer communication pathways between the nucleoplasm and cytoplasm. The pathways are restricted to so-called transport receptors, proteins that taxi molecules into and out of the nucleus. If a molecule wishes to enter or leave the nucleus, it associates with a transport receptor. The complex passes through the pore and then dissociates. The question is why transport receptors can pass the nuclear pores while other proteins cannot. The answer lies in the role of FG-repeat proteins lining the pores and filling much of their free volume. These proteins are disordered peptides, consisting of repeating phenylalanine-glycine (FG) residues separated by a sequence of hydrophilic linker residues. Only proteins that interact favorably with the FG-repeat regions can pass through, while other proteins are excluded. A recent report used molecular dynamics via NAMD to examine the way in which the transport factor NTF2 interacts with the FG-repeats. The study described binding spots for FG-repeat peptides on the surface of NTF2, confirming known binding spots discovered previously via experimental means, and suggesting the existence of further binding spots. The new binding spots may play a role in steering NTF2, upon import or export, along a particular path through the nuclear pore. See also a previous highlight from January 2006, "Gateway to the Nucleus", as well as our webpage on the nuclear pore complex.
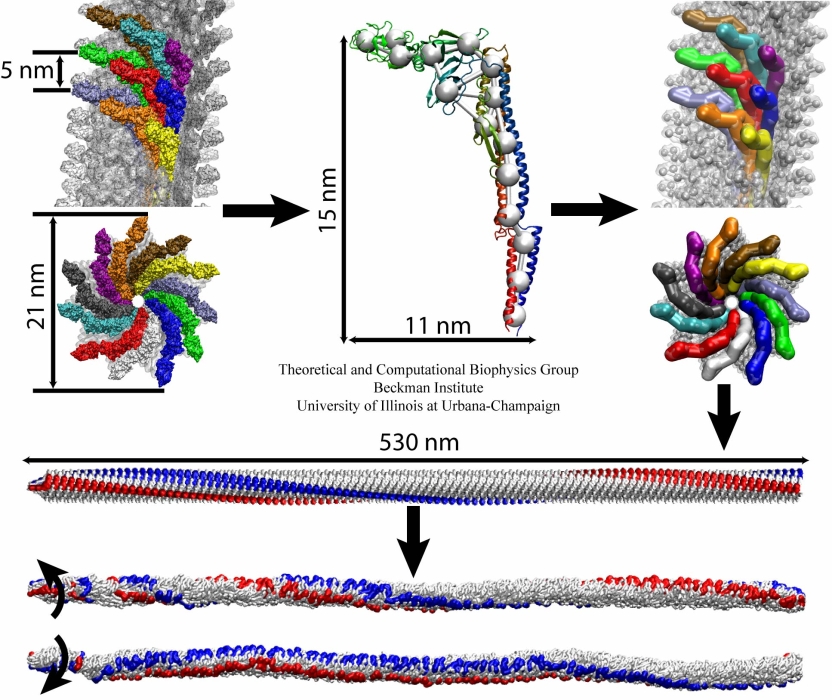
image size:
299.9KB
made with VMD
The bacterial flagellum is a large biomolecular assembly used by many types of bacteria as a helical propeller for forward swimming and turning. The flagellum is remarkable in that its properties differ greatly depending on the direction in which it is rotated, allowing the bacterium to switch between swimming straight ("running") and turning ("tumbling"). The mechanics of the flagellum are of interest both to biologists and mechanical engineers. The molecular mechanisms of the transition in the flagellum between running and tumbling modes is unknown. Because of the flagellum's size (several micrometers in length) and composition (made up of 30,000 protein subunits) it presents a challenge to computational modeling. Researchers have now achieved an advance describing the flagellum in both its running and tumbling state. For this purpose, the researchers developed a computational model of the system that glosses over atomic level detail, but resolves the shapes of all proteins making up a bacterial flagellum, simulating a simplified version of the system using the program NAMD. The results, reported recently, showed that the flagellum's transition between swimming straight and tumbling is triggered by friction due to the water around the bacterium. More information on the flagellum project can be found here.
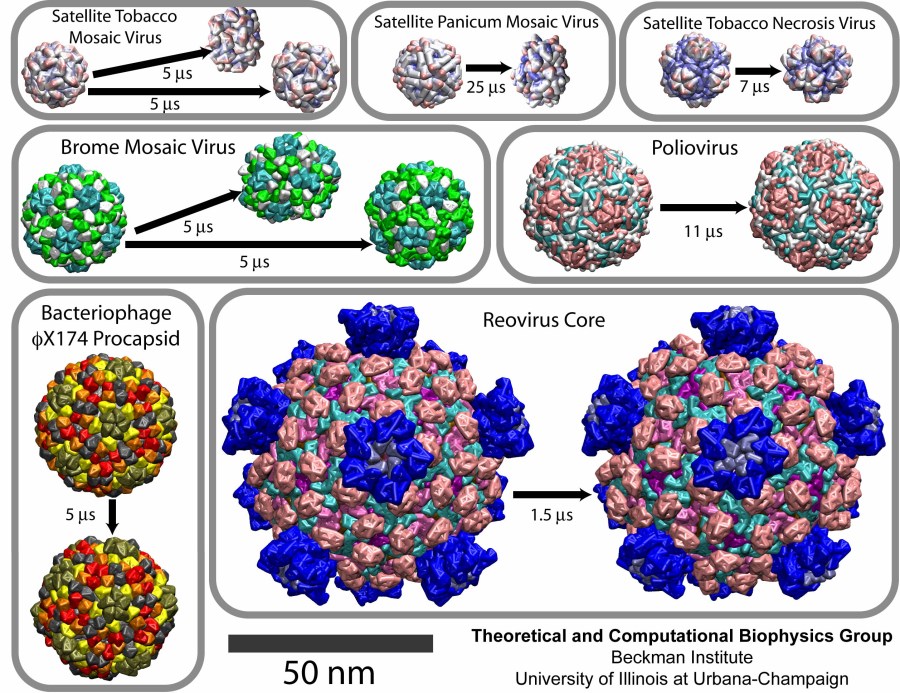
image size:
219.5KB
made with VMD
Viruses are the cause of many human diseases, from the common cold to AIDS, and medicine is continuously searching for better ways to battle viruses through vaccination or medication. Detailed knowledge of the life cycles of viruses should be useful in the treatment of viral diseases. A key focus of investigations is the virus capsid, a protein coat that protects the viral genome, but also triggers release of the genome and other viral factors upon contact with the body's cells. X-ray crystallography has resolved the average structures of many types of virus capsids, providing the basis for detailed investigations, for example by means of molecular dynamics methods, of capsid dynamical properties, e.g., in assembly and disassembly. Unfortunately, due to their large size most virus capsids are beyond the reach of molecular dynamics simulations, with one notable exception (see the March 2006 highlight "Simulating an Entire Life Form"). This earlier simulation allowed researchers to develop and test a method for coarse-grained molecular dynamics simulations that glosses over atomic detail and, thereby, permits microsecond descriptions of entire viral particles. As reported recently (see also journal cover) such simulations, employing the program NAMD, were applied to the empty capsids of several viruses. These simulations revealed a variety of behaviors, from rapid collapse to high stability, depending on the strength of interactions between the proteins from which capsids are built. The new method offers unprecedented views of capsid dynamics that may assist in battling viral diseases. More information on the simulations can be found on our virus web page.
image size:
91.7KB
made using VMD
Bionanotechnology involves a marriage of two different materials: inorganic solids, like silica, and biomolecules, like DNA. The new combinations of materials have to be mastered on the laboratory bench as well as in computer simulations. On the bench, devices are manufactured and tested, in the simulations, they are imaged and designed. So far inorganic solids and biomolecules were simulated successfully, but only separately. To join the materials requires as much effort in simulations as on the bench. Even just the interaction of inorganic solids (like silica) with physiological solutions (water and ions) demands challenging descriptions of silica surface properties. As reported recently, researchers have now succeeded in describing accurately the wetting (by water) of amorphous silica, an essential material for nanoelectronics, clearing a major hurdle to simulating bionanotechnological devices, for example, those suggested for rapid and economical sequencing of DNA (see also Nov 2005 and Oct 2004 highlights). More on silica-water interaction here.
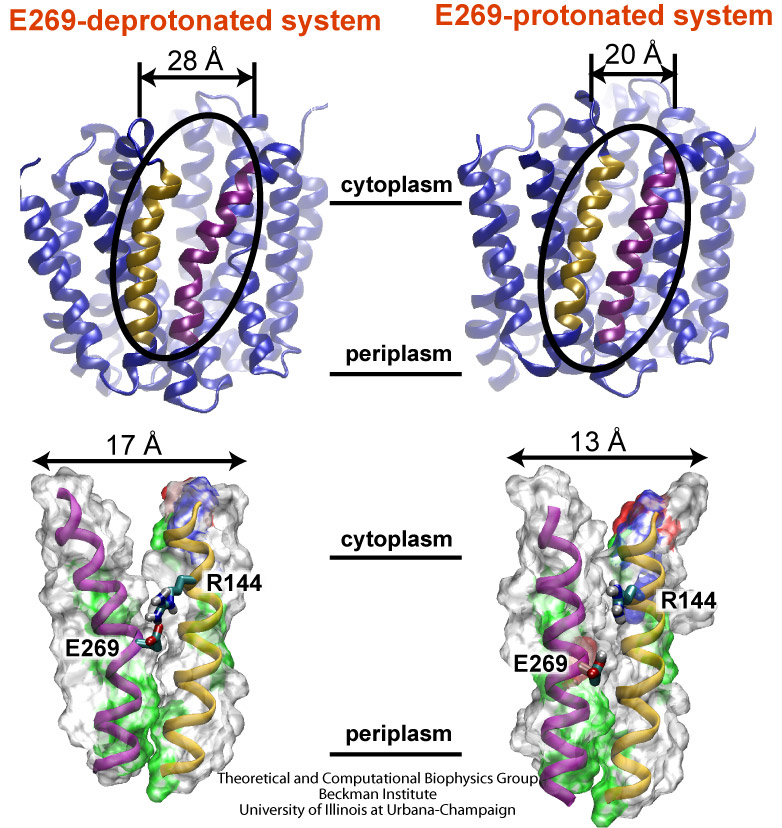
image size:
215.8KB
made with VMD
Escherichia coli are bacteria living in the intestines of mammals as part of their healthy gut flora, but also causing disease outside of the gut. The bacteria import from their environment nutriments, for example molecules of lactose, a sugar. For this purpose Escherichia coli employs in its cell membrane a protein channel, lactose permease, that translocates the sugar outside-in. This is the bacterium's "sweet tooth". To establish the unidirectional sugar transport, the bacterium utilizes an electrical potential maintained in the form of a trans-membrane proton gradient (more protons on the outer cellular than on the inner cellular side of the membrane). Protons, very small ions, that enter the channel from the outside one at a time, open the outer channel entrance. This permits access of lactose that gets bound inside the channel. Release of the proton to the cell interior closes the outer channel entrance and opens the inner channel entrance, such that the bound lactose can enter the cell. Despite extensive and elegant biochemical studies, the physical mechanism that couples unidirectional proton and sugar translocation is not yet known in detail. A crystallographic structure of lactose permease permitted now investigations into this mechanism by means of molecular dynamics simulations using NAMD. The simulations, reported in a recent publication, showed one step of the proton - sugar translocation, namely how binding and unbinding of the proton activates a spring-like bond, a so-called salt bridge, that closes and opens the inner channel exit. More information on the lactose permease project can be found here.
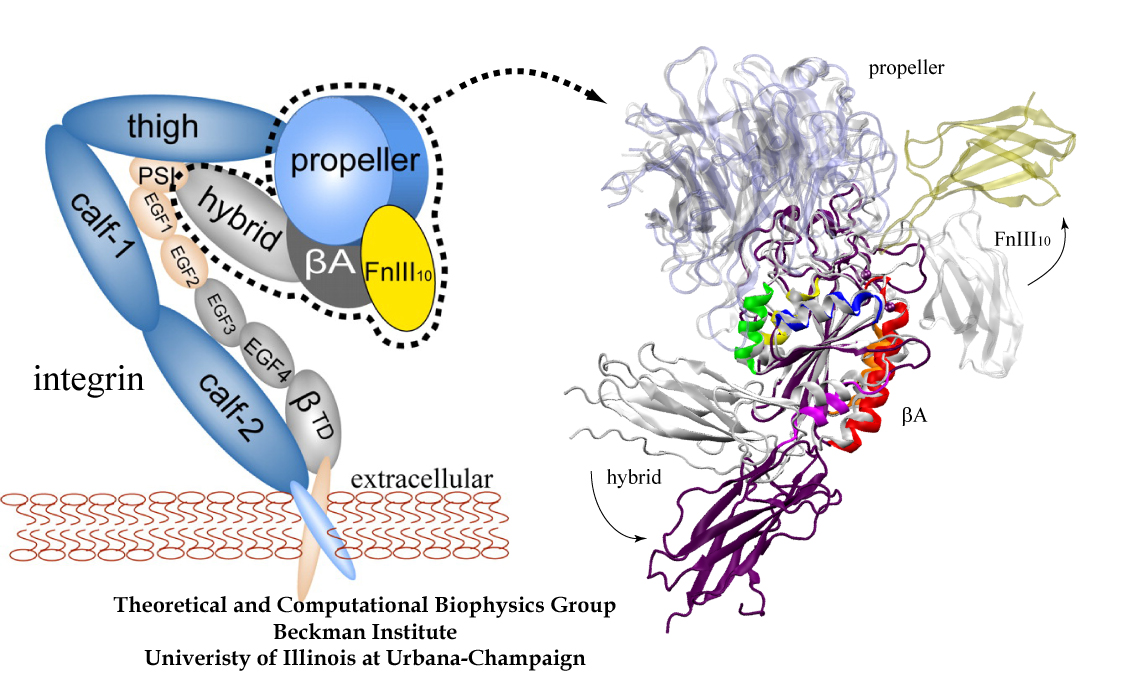
image size: 468.6KB
Mammalian cells adhere to each other forming tissues. The adhesion is due to a network of proteins, so-called extracellular matrix proteins, "gluing" the cells together. The cell membranes are too soft to provide anchoring points for the extracellular matrix proteins; rather, the cells furnish on their outer surface specialized hooks for anchoring the extracellular matrix proteins. The hooks, in the form of surface proteins, are linked directly through the membranes to the intracellular cytoskeleton that stabilizes and shapes cells. Integrins are an important family of such surface proteins that form hooks specific for certain types of extracellular matrix proteins. The hooks are flexible, they can be open for contacts or closed, the switch being induced by signals from inside or outside the cell through interactions with other proteins. The interactions between integrins and extracellular matrix proteins are rather complex, as the proteins are composed of many subunits; fortunately, their overall structures are presently being solved through crystallography. In a recent report a major component of an integrin and an extracellular matrix protein have been investigated through molecular modeling using NAMD, including steered molecular dynamics. The study described in detail how the extracellular matrix protein induces a transition in integrin, potentially strengthening its adhesion property. See also previous highlights: the May 2006 "Killer's Entry Route", Dec 2004 "Snap Fastener on Biological Cells", Dec 2003 "Body's Glue", and Mar 2002 "Cells Sense Push and Pull". More on modeling of extracellular matrix proteins and integrins can be found here.
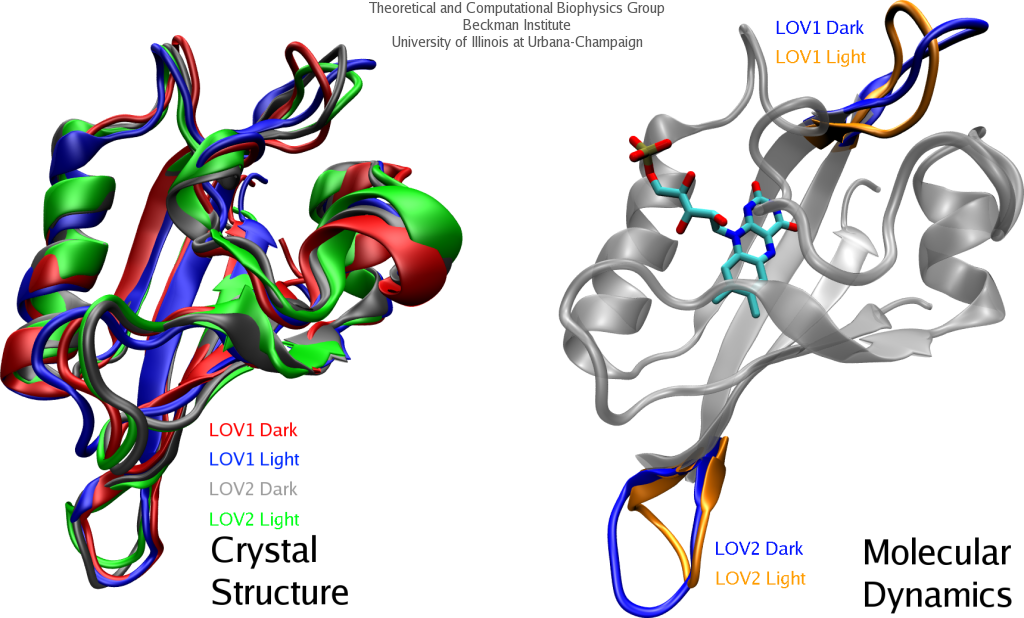
image size:
397.0KB
made using VMD
Most forms of life need to detect and respond to changes in their environment for survival and optimal growth. For this purpose organisms rely on receptors that are based on sensory proteins. In plants, several sensory proteins detect the ambient light for optimal exposure of their photosynthetic apparatus. One class of plant light sensors, the phototropins, influence photosynthesis and induce the transition between root and stem growth when seedlings emerge out of the ground. Induction is activated through several protein domains, two of which actually absorb light and for their sensitivity to light, oxygen, and voltage, are called LOV1 and LOV2 domains. Understanding the LOV domains' involvement in activation is important for studying the signaling mechanisms of other types of sensory proteins. Strangely, light absorbed by LOV domains is observed to lead to a distinct, but only very minute, structural change that does not explain how activation might come about. NAMD-based molecular dynamics simulations of the LOV domain have now revealed, as reported in a recent publication, that photoactivated LOV domains exhibit altered patterns of motion that can induce a signal for plant cells. More information may be found on our biological photoreceptors website.
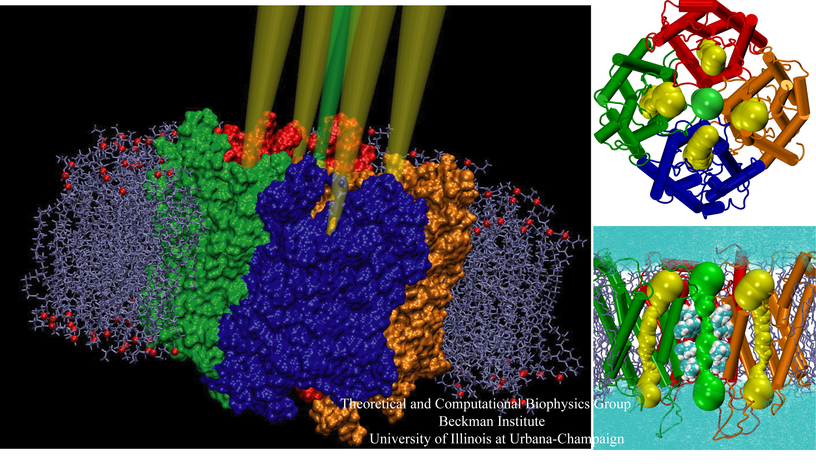
image size:
501.0KB
made using VMD
Sometimes analogies go a long way, surprisingly long. Aquaporins are ubiquitous water channels in living cells, known to be tetrameric, each unit contributing one pore. This much is certain and this is where an analogy begins, namely with a British spy ring that passed information to the Soviet Union during world war II and into the 1950s. The ring is often referred to as the Cambridge Four since the spies, when recruited, were undergrads at Cambridge Trinity College and there were four of them (cryptonyms Stanley, Homer, Hicks, and Johnson). But a Fifth Man was long suspected, yet never formally identified. Here the analogy continues: aquaporin was suspected to sport a fifth pore, supposedly at its center, where its four subunits join (hence known as the tetrameric pore). Strong, but not yet completely conclusive, evidence has now been put forward in a recent report that the central pore, actually quite plainly visible to the eye when aquaporins are inspected by molecular graphics, e.g., with VMD, is an ion channel gated by a common cellular signaling molecule, cGMP. The evidence stems from a combined computational (molecular dynamics using NAMD) and experimental (verifying computationally suggested mutants) study. More information on the five pores can be found on our aquaporin website, more on the Cambridge Five here. But the analogy goes further. Today it is suspected that the Cambridge Five actually had more than five members and the same holds for the pores of aquaporin. An ongoing investigation has lead to evidence that the further pore members conduct gases, for example carbon dioxide. Hopefully, we will know one day with certainty all members of the Cambridge Five and all pores of aquaporin.

image size:
285.6KB
made using VMD
The computer processor is the workhorse of biomolecular modeling, with NAMD the plow to which a single processor or a team of thousands may be hitched. The recent release of NAMD 2.6 has extended the drawbar to harness the power of several thousand processors: 2000 on a Cray XT3 and 8,000 on an IBM Blue Gene/L. This permits the efficient simulation of an entire ribosome, the cell's protein factory, comprising 3,000,000 atoms when solvated. But the features and increased performance of NAMD 2.6 are also available to the scientist with only a laptop, on which a domain of the muscle protein titin (10,000 atoms solvated) can be readily simulated. NAMD has also become more versatile, supporting more force fields (OPLS, CHARMM with CMAP cross terms), calculating free energies, and executing customizable replica exchange simulations. In addition, NAMD can now be called from the structure analysis program VMD to calculate, for example, interaction energies between protein domains. Like increased horsepower in transportation, increased simulation power opens new routes, routes to study entire systems of biopolymers like the ribosome, not just one piece.
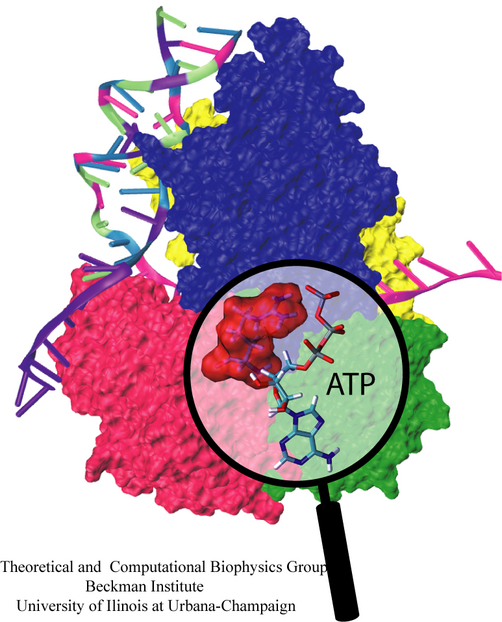
DNA with its famous double helix structure stores the genetic information of all life forms known. In order that this information is read, the double helix needs to be first unwound and separated into single helices or strands. This is achieved by cellular motor proteins called helicases that operate on already separated DNA strands. The helicases specialize in unwinding and separating the DNA double helix by scooting along one of DNA's single strands against the point where the two strands merge into the double helix; pushing against this point unwinds and separates the double helix further. The helicases are driven by energy stored in molecules of ATP which bind to the protein and get released in their so-called hydrolyzed, lower energy, form. Based on atomic resolution structures, researchers have studied now one of the smallest helicases known, PcrA, from the electronic to the functional level carrying out quantum mechanical/molecular mechanical simulations (as described in a first publication), as well as a combination of classical molecular dynamics simulation, using NAMD, and stochastic modeling calculations (described in a second publication and a third publication). This resulted in an overall explanation of how ATP's hydrolysis powers helicase activity which has been reported in a fourth publication. The researchers discovered that PcrA moves with two "hands" along single stranded DNA; when ATP binds, one "hand" moves along the DNA; when ADP and Pi (the hydrolysis products of ATP) unbind, the other "hand" moves; through a molecular "trick" both "hands" move in the same direction. Amazingly, the hand movement arises mainly from an increase in random mobility of the hands. i.e., is not enforced. Physicists refer to the underlying mechanism as a ratchet mechanism that was indeed long suspected to drive molecular motors. Interestingly, the helicase motor is very closely related to a wide class of other biological motors, for example the FoF1-ATP synthase (see Mar 2004 and Nov 2004 highlights). For more information visit our helicase research website.
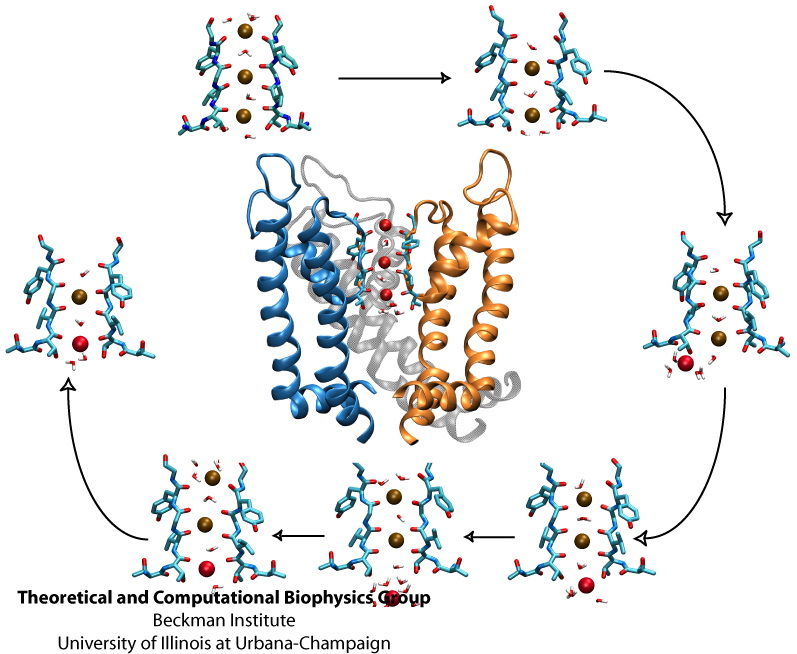
Biological cells, in particular neurons, maintain an inside-outside voltage gradient through active transport of ions (Na+, K+, Cl-, and others) across their membranes. The flow of the ions down their gradients through membrane channels is highly selective for each ion. The high selectivity permits nerve cells to signal each other through voltage spikes, which are produced through transient changes of channel conductivities for Na+ ions (channels open and close in about a ms) and K+ ions (channels open and close in about 10 ms). Crucial for the generation of voltage spikes is the selective, yet quick, conduction of ions, but as one knows from personal experience at border crossings, high selectivity and quick crossing seem to be mutually exclusive. Yet biological ion channels reconcile selectivity and speed. Prior experimental work, primarily that of year 2003 Nobelist MacKinnon, as well as computational work suggested how potassium channels achieve selectivity and speed. But until recently no high resolution atomic structure of a potassium channel was known in the open form and the suggested mechanism could not be tested under natural conditions through atomic level simulations. Last year's solution of the structure of the potassium channel Kv1.2 in its open form made it finally possible to simulate, using NAMD, the conduction of ions through Kv1.2 driven by a voltage gradient. The results reported recently confirmed indeed the high selectivity - high speed mechanism suggested earlier, namely a billiard-type motion of two and three ions, the last ion kicking the first ion out. The simulations revealed for the first time, through movies, the overall permeation process, including the jumps of ions between energetically favorable binding sites and the sequence of multi-ion configurations involved in permeation. More on our potassium channel web site.
image size:
249.5KB
made with VMD
How far and how well molecular biologists can look into the living cell depends as much on microscopes and observations, as it depends on computers and their software. The premiere software for looking into the molecular world of the cell, VMD, has made a big leap forward in broadening the molecular horizon of life scientists through its new release, VMD 1.8.5. Researchers are offered now a fresh view through a modern unified bioinformatics environment, MultiSeq, combining sequence and structure analysis for proteins and amino acids. VMD, now literally more colorful, lets scientists quickly exchange VMD views through integration of BioCoRE, calculate APBS electrostatics maps, call on NAMD to calculate energies, build and mutate structures, determine easily force field parameters, and navigate through proteins with a flying camera. VMD 1.8.5, though only a minor version number different from the previous release, includes now many new structure building and analysis tools that make it easier for modelers to set up, run, and analyze computer simulations of biomolecules.
Many proteins interact with gas molecules such as oxygen to perform their functions. In most cases, the gas molecules must reach sites buried deep inside the proteins that bind the molecules, with no obvious way in. Understanding how, for example, oxygen enters the protein, and mapping out which pathways it takes has been a long-standing challenge. As reported recently, computational biologists, inspired by previous work on the hydrogenase enzyme (see the September 2005 highlight), have developed a method, called implicit ligand sampling, that maps the pathways taken by gas molecules inside proteins. The mapping is determined by monitoring fluctuations of the protein, surprisingly, in the absence of the gas molecules. The mapping method is available in the most recent version of the program VMD used for structure and sequence analysis of proteins. The researchers applied the method to myoglobin, an oxygen-storing protein present in muscle cells, and determined detailed three-dimensional maps of oxygen and carbon monoxide pathways inside the protein (for more information see our web page). While some details of these pathways were already known from experiment, the implicit ligand maps revealed a large number of new pathways and suggest that oxygen enters myoglobin using many different entrance doors.
image size:
300.4KB
made with VMD
The living state of biological cells manifests itself through mechanical motion on many length scales. Behind this motion are processes that generate and transform mechanical forces of various types. As with other cell functions, the machinery for cellular mechanics involves proteins. Their flexible structures can be deformed and restored, and are often essential for handling, transforming, and using mechanical force. For instance, proteins of muscle and the extracellular matrix exhibit salient elasticity upon stretching, mechanosensory proteins transduce weak mechanical stimuli into electrical signals, and so-called regulatory proteins force DNA into loops controlling, thereby, gene expression. In a recent review, the structure-function relationship of four protein complexes with well defined and representative mechanical functions has been described. The first protein system reviewed is titin, a protein that confers passive elasticity on muscle. The second system reviewed is the elastic extracellular matrix protein fibronectin and its cellular receptor integrin. The third protein system covered are the proteins cadherin and ankyrin involved in the transduction apparatus of mechanical senses and hearing. The last system surveyed is the lac repressor, a protein which regulates gene expression by looping DNA. In each case, molecular dynamics simulations using NAMD provided insights into the physical mechanisms underlying the associated mechanical functions of living cells. (more on our mechanobiology web site).

In a recent paper experiences designing and deploying NAMD-G, an infrastructure for executing biomolecular simulations using the parallel molecular dynamics code NAMD within the context of a Computational Grid, are described. The NAMD-G project, and associated paper, is the result of a collaboration between the Theoretical and Computational Biophysics Group (TCBG) and the National Center for Supercomputing Applications (NCSA). James Phillips, Senior Research Programmer for NAMD, and Jordi Cohen, a Ph.D. candidate in Physics at the University of Illinois at Urbana-Champaign, contributed on behalf of the TCBG, while Richard Kufrin, Senior Research Programmer, and Michelle Gower, Research Programmer, led development for NCSA.

NAMD (NAnoscale Molecular Dynamics) can simulate the movement of proteins with millions of atoms, making it the world's fastest parallel molecular dynamics program. The NAMD development team will continue to incorporate the latest parallel-computing advances into NAMD, which already runs efficiently on several thousand parallel processors. Pictured to the right is the current development team for NAMD. James Phillips, a PhD in Physics, is Senior Research Programmer for NAMD at the Theoretical and Computational Biophysics Group (TCBG). Dr. Laxmikant Kale, Professor of Computer Science at UIUC, is a Co-PI at TCBG and leader of the Parallel Programming Laboratory. Kale's computer science graduate students, Chee Wai Lee, Chao Mei, and David Kunzman, also contribute to various aspects of NAMD development.

Many proteins store gases like oxygen, carbon dioxide, and nitric oxide, or react with them. The gases are conducted into the protein through access routes that exist only in passing and as a result of a protein's fluctuations. Accordingly, access routes are difficult to establish, but researchers are now able to image gas access pathways inside proteins computationally. The new method has many implications for biotechnology and science (see our hydrogenase page and Sep 2005 highlight, "Hydrogen Fuel from Protein"). Imaging gas access systematically over whole protein families, e.g., the family of myoglobins, requires a large number of calculations that need to be run and monitored. The traditional means of doing so is very wasteful of the researchers' time. To solve this problem, NAMD-G, a grid-based automation engine for biomolecular simulations running the NAMD software, has been developed in a collaboration with the National Center for Supercomputing Applications (see recent paper). From the researchers' workstations, NAMD-G "farms out" a large number of calculations, in parallel, to supercomputers on the TeraGrid. NAMD-G monitors and manages multiple sequences of calculations at distant sites, and performs the necessary data transfers and backups on an as-needed basis. While the gas transport simulations provide a clear scientific driver for the development, NAMD-G is quite general and will aid any NAMD user with access to the TeraGrid. The result? Less time spent baby-sitting runs and more time for science.

For hard-working scientists, the task of maintaining a single desktop computer is an unwelcome distraction. But what if your work requires the power of ten or a hundred machines? Our recent series of workshops (Sep 2005, Nov 2005, Mar 2006, and Apr 2006) has given nearly one hundred participants hands-on experience installing and using low-cost Linux clusters. Students were taught to eliminate many sources of complexity, such as hard drives, and to automate what remained with cluster management software and a queueing system. Lectures on cluster design stressed the importance of knowing which applications would be run and choosing cost-effective hardware to meet those specific needs, as well as less-obvious aspects of cluster acquisition such as electrical power, cooling, and the purchasing process. After assembling and installing small four-node clusters, students ran both the molecular dynamics program NAMD and a more typical parallel application that they compiled from scratch. Most participants were motivated by concrete plans to build clusters for their own groups in the near future and felt better-equipped to do so following their experience.
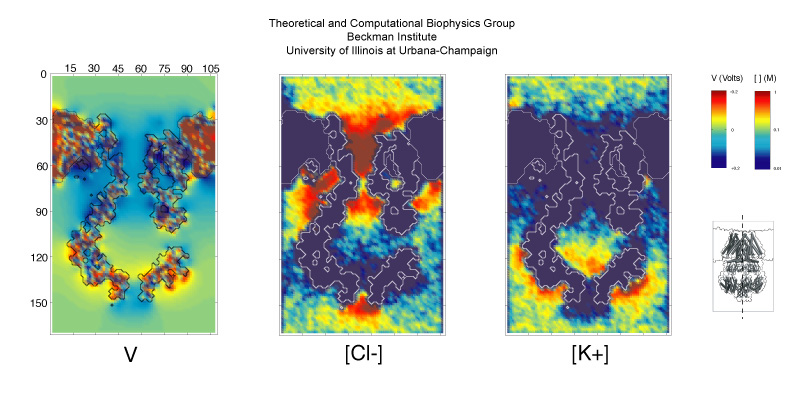
image size:
112.7KB
Bacterial cells, like those of Escherichia coli, protect themselves against sudden inside-out pressure differences that arise osmotically from changes in a cell's environment and that could burst the cellular envelop. The protection is achieved through so-called mechanosensitive channels in the cell membrane. One such channel, that dissipates like a safety valve pressure differences across the Escherichia coli cell membrane, is contributed by the protein MscS. Upon tension in the cell membrane, that can also be applied systematically in the laboratory, the channel opens and permits molecules to pass, as best measured through an ion current leaking through the stretched membrane. MscS is a channel with a balloon-like filter, the function of the latter being still a mystery (see Nov 2004 highlight, "Japanese lantern protein"). Now computational biologists using NAMD teamed up with device engineers using BioMoca to study MscS as reported recently. The team monitored the mysterious MscS computationally over several microseconds, a record time for protein simulations. MscS was found to permit water passage, but to also exhibit strong electrostatic forces that focus ions streaming through its filter balloon and channel. This suggests MscS to be both a hydrostatic and an electrical safety valve. Even though now better known, MscS' entire function remains shrouded in mystery (more on our MscS web site).
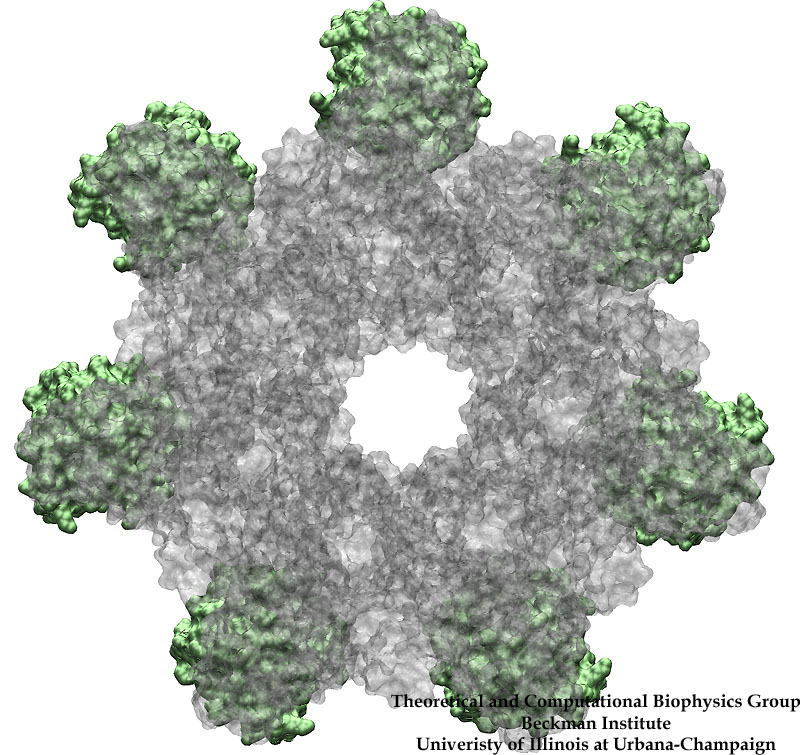
image size:
194.1KB
made with VMD
Bacillus anthracis, the cause of anthrax, is one of the most lethal bacteria. In addition to its ability to infect animal and human cells, the bacterium attacks also the cells of the host's immune system, the so-called macrophages. For this purpose the anthrax bacterium releases three types of proteins, or toxins, into the blood stream of the host: protective antigen, lethal factor, and edema factor, referred to as PA, LF, and EF, respectively. LF and EF team up with PA, which transports them into a host macrophage cell. Once inside the cell, LF converts ATP to cyclic AMP, while EF disables MAPKKs, a family of signaling proteins. These attacks disrupt various cellular signaling pathways of macrophages and some other cells, essentially shutting down the host's immune system and often leading to death of the host. To invade macrophages, the toxins take an intricate entry route that involves binding to a cell receptor, capillary morphogenesis protein 2 (CMG2), inducing the cell to internalize the toxins in a bubble like membrane (endosome), the bubble wall being then punctured by seven PAs forming a channel upon a chemical (acidifying) trigger from the host; the channel permits then their lethal cargo, LFs and EFs, to slip into the cell. How exactly the PAs punctured the endosome wall remained a mystery. In a recent report the entry route has been resolved now in greater detail through molecular dynamics simulations using NAMD. The report reveals how acidic conditions in the endosome trigger conformational changes of the PA complex necessary for pore formation, and provides structural insights into the role of unusual interactions between the PAs and its receptor CMG2. Visit also our anthrax toxin webpage.

image size: 87.6KB
Computational tools, like the molecular graphics program VMD and molecular dynamics program NAMD, move rapidly from theoretical to experimental biology. To train researchers in the proper use of computational tools, a series of hands-on workshops was organized in the US and Australia in 2003-2005 (see July 2005 highlight). This year the first European hands-on workshop started a new generation of training with three novel features. First, the workshop addressed mainly bench scientists in need of computational methods. Second, the workshop introduced a key expansion of VMD that turned a mainly structurally oriented visualization program into a structure and sequence analysis program. This is achieved through a multiple sequence analysis tool in VMD, called multiseq. Third, all training material has been extended to multiple platforms and participants could bring their own laptops for the training sessions. As in the previous series, participants enjoyed workshop lectures that introduced concepts and good uses of biocomputing software, but were most enthusiastic about practical tutorials that provided opportunities to learn by example and to apply newly mastered tools to their own research. The participants carried all lecture material and software home on a DVD; others can obtain the same material through our web site (workshop lectures, tutorials, case studies, VMD, NAMD).
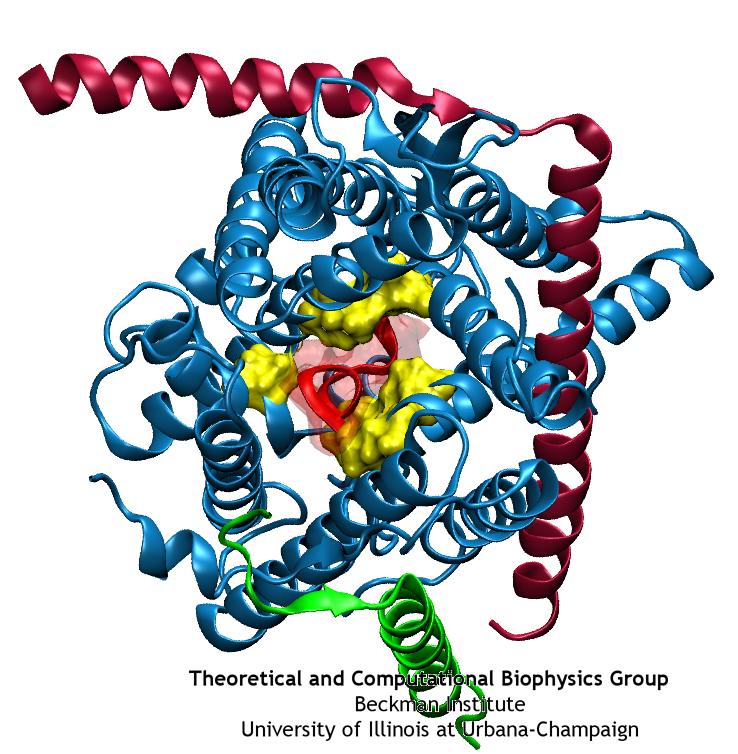
image size:
100.6KB
made with VMD
Anyone who has attempted to fit a long piece of thread through a needle's eye realizes how difficult fitting something so small and flexible into such a small hole can be. Yet this action is carried out every second in every living cell. Flexible polypeptides, proteins, often have to cross a cellular membrane to get to their correct location, whether that location is an organelle within the cell or even outside of it. To accomplish this, they are pushed through a protein pore in an unfolded conformation much like a long string. The channel that accepts the string-like proteins, the protein translocon, allows only certain proteins to pass, while restricting access to molecules even much smaller than the macromolecular proteins. As reported in a recent publication, computer simulations using the molecular dynamics program NAMD helped to answer the question of how such a small channel could achieve this feat, demonstrating how the channel itself can be flexible yet resilient during a protein-crossing event and also elucidating in part how it can maintain such tight control over what is permitted to cross. For more information, see our Protein Translocation website.
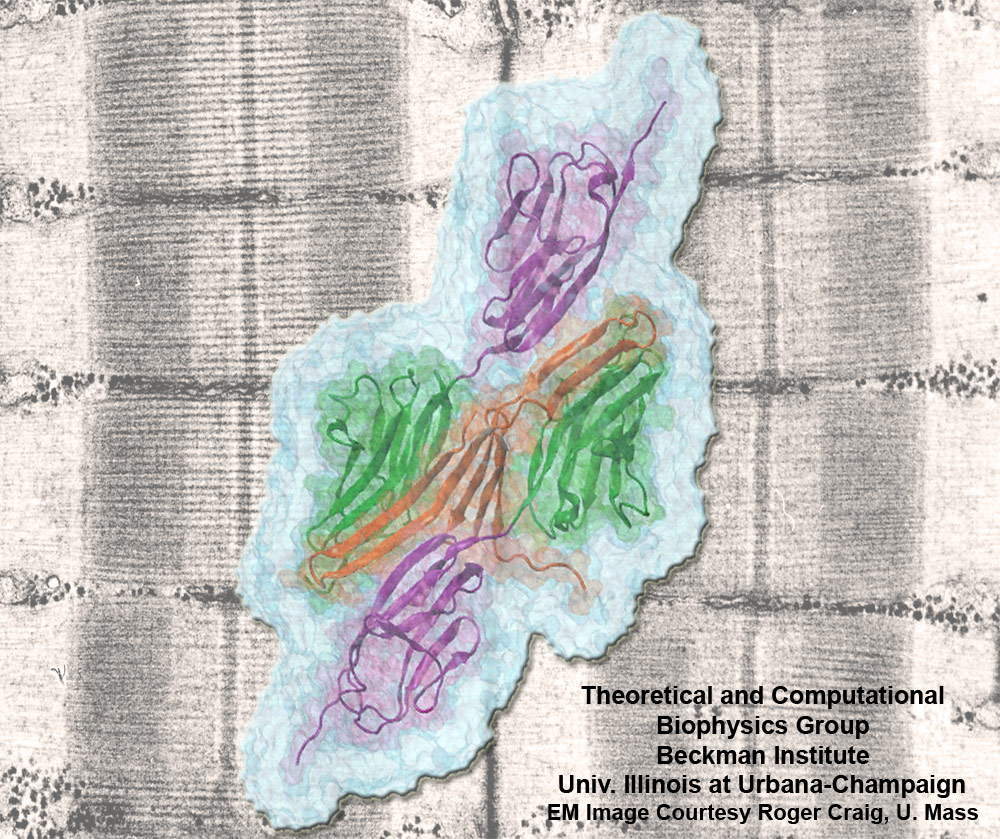
image size:
301.5KB
made with VMD
Muscle fibers, through their so-called thick and thin filaments, contract and extend in doing their work. To render the fibers elastic and protect them from overstretching, the thick filaments are connected through a long and thin elastic protein, titin, to the base of the fibers. Titin, by far the longest protein in human cells, is a molecular bungee cord and, like such cord, must be affixed firmly to the base. How this is done was a mystery until crystallographers took the first atomic resolution image of the system: it turns out that two titins are spliced together at their ends like ropes. The splicing involves a third small protein, the titin-telethonin-titin system forming a U. The U apparently is thrown over a bollard-like cellular structure to hold the thick filaments much like boats are held by bollards and ropes at their mooring place. The crystallographers teamed up with computational biologists to investigate the mechanical strength of the titin - telethonin - titin cord by means of molecular dynamics simulations using NAMD. As reported recently, the cord has great mechanical strength due to an extended network of hydrogen bonds between beta-strands, common structural features in proteins, that in the present case form a sheet extending through all three proteins. This discovery explains how living cells can splice cellular proteins together through a system of hydrogen-bonded beta-strands that extend through several proteins. Interestingly, such beta-strands were seen previously in cases of diseases like Alzheimers where the feature leads, however, to pathological assembly of proteins. What needs to be understood now is how the telethonin glue is applied only to the right spots in the cell and how the cells prevent telethonin from splicing together the wrong proteins. For more information visit our titin-telethonin web page.
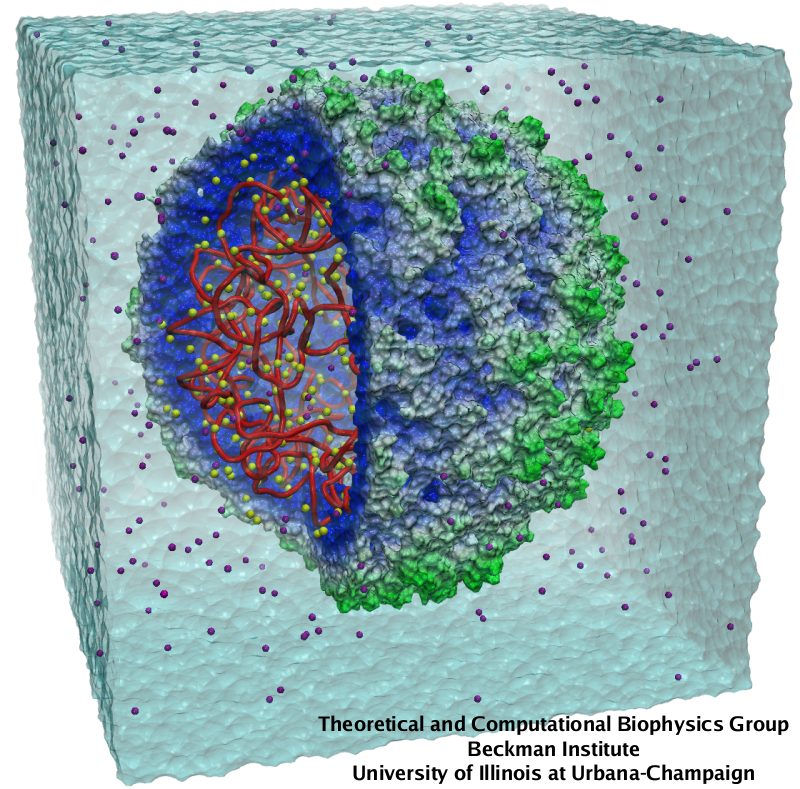
image size:
150.4KB
made with VMD
Viruses, the cause of many diseases, are the smallest natural organisms known. They are extremely primitive and parasitic such that biologists refer to them as "particles", rather than organisms. Viruses contain in a protein shell, the capsid, their own building plan, the genome, in the form of DNA or RNA. Viruses hijack a biological cell and make it produce from one virus many new ones. Viruses have evolved elaborate mechanisms to infect host cells, to to produce and assemble their own components, and to leave the host cell when it bursts from viral overcrowding. Because of their simplicity and small size, computational biologists selected a virus for their first attempt to reverse-engineer in a computer program, NAMD, an entire life form, choosing one of the tiniest viruses for this purpose, the satellite tobacco mosaic virus. As described in a recent report, the researchers simulated the virus in a small drop of salt water, altogether involving over a million atoms. This provided an unprecedented view into the dynamics of the virus for a very brief time, revealing nevertheless the key physical properties of the viral particle as well as providing crucial information on its assembly. It may take still a long time to simulate a dog wagging its tail in the computer, but a big first step has been taken to simulate living organisms. Naturally, this step will assist modern medicine (more on our satellite tobacco mosaic virus web page).
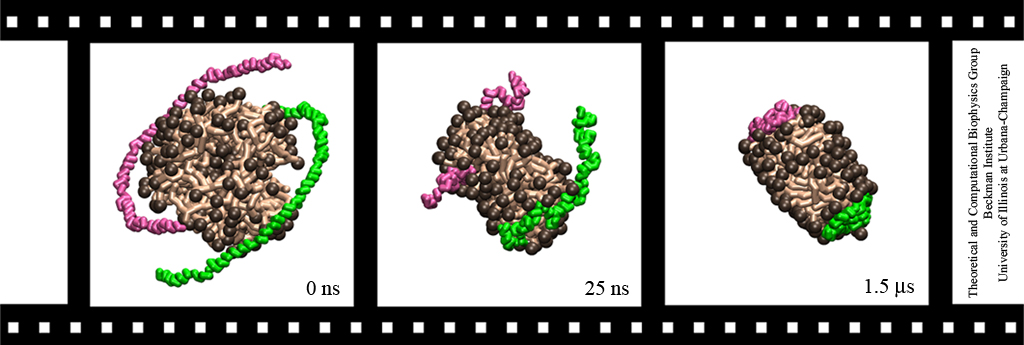
image size:
153.6KB
made with VMD
Lipoproteins are protein-lipid particles which circulate in the blood collecting cholesterol, fatty acids, and lipids. Low levels of one such lipoprotein particle, called high-density lipoprotein (HDL) or "good cholesterol", has been implicated in the increased risk of coronary heart disease. The ability of lipoproteins to transport lipid and cholesterol through the blood is amazing since these types of particles are not generally soluble in blood plasma. However, when HDLs assemble, proteins wrap themselves around the lipids and cholesterol, shielding the lipid tails from the aqueous environment. Native HDL exhibit a variety of shapes and sizes, for example forming a discoidal particle. Conventional high-resolution imaging techniques, such as NMR and X-ray crystallography, cannot resolve how lipid and cholesterol are being accommodated by HDL, but the assembly and geometry of HDL discs can be captured using computer simulations. Unfortunately, the long time scales required for HDL assembly was a major stumbling block. Now a new simulation method, coarse-grained modeling in conjunction with the molecular dynamics program NAMD, has permitted the simulation of HDL assembly as recently reported. The simulations show that lipids quickly aggregate into a bilayer from their initial spherical "micelle" shape and that the two proteins subsequently attach to either side forming a belt surrounding the lipid core. For more information see HDL & nanodisc and coarse-grained modeling.
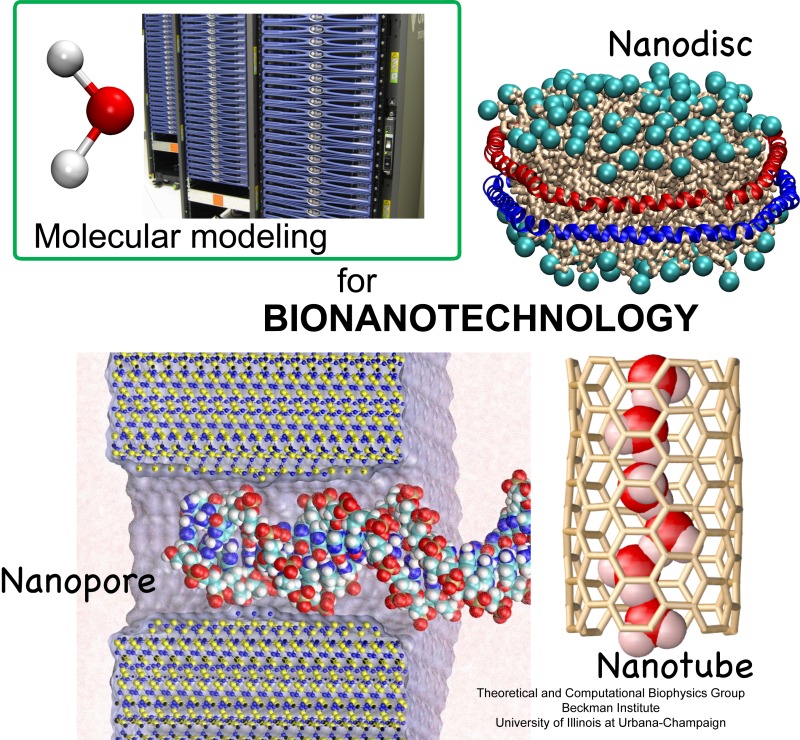
image size:
225.3KB
made with VMD
Molecular modeling with NAMD (NAnoscale Molecular Dynamics) promises to become a key methodology for research and development in bionanotechnology. Molecular modeling provides nanoscale images at atomic and even electronic resolution, predicts the nanoscale interaction of yet unfamiliar combinations of biological and inorganic materials, and can evaluate strategies for redesigning biopolymers for nanotechnological uses. The methodology's value has been reviewed for three uses in bionanotechnology. The first involves the use of single-walled carbon nanotubes as biomedical sensors where a computationally efficient, yet accurate description of the influence of biomolecules on nanotube electronic properties and a description of nanotube - biomolecule interactions were developed; this development furnishes the ability to test nanotube electronic properties in realistic biological environments (see Dec 2005 highlight). The second case study involves the use of nanopores manufactured into electronic nanodevices based on silicon compounds for single molecule electrical recording, in particular, for DNA sequencing. Here, modeling combining classical molecular dynamics, material science, and device physics, describes the interaction of biopolymers, e.g., DNA, with silicon nitrate and silicon oxide pores, furnishes accurate dynamic images of pore translocation processes, and predicts signals (see Nov 2005 and Oct 2004 highlights). The third case involves the development of nanoscale lipid bilayers for the study of embedded membrane proteins and cholesterol. Molecular modeling tested scaffold proteins, redesigned lipoproteins found in mammalian plasma that hold the discoidal membranes in shape, and predicted the assembly as well as final structure of the nanodiscs (see Feb 2005 highlight). In entirely new technological areas like bionanotechnology qualitative concepts, pictures, and suggestions are sorely needed; the three exemplary applications document that molecular modeling can serve as a critical "imaging" method for bionanotechnology, even though it may still fall short on quantitative precision.
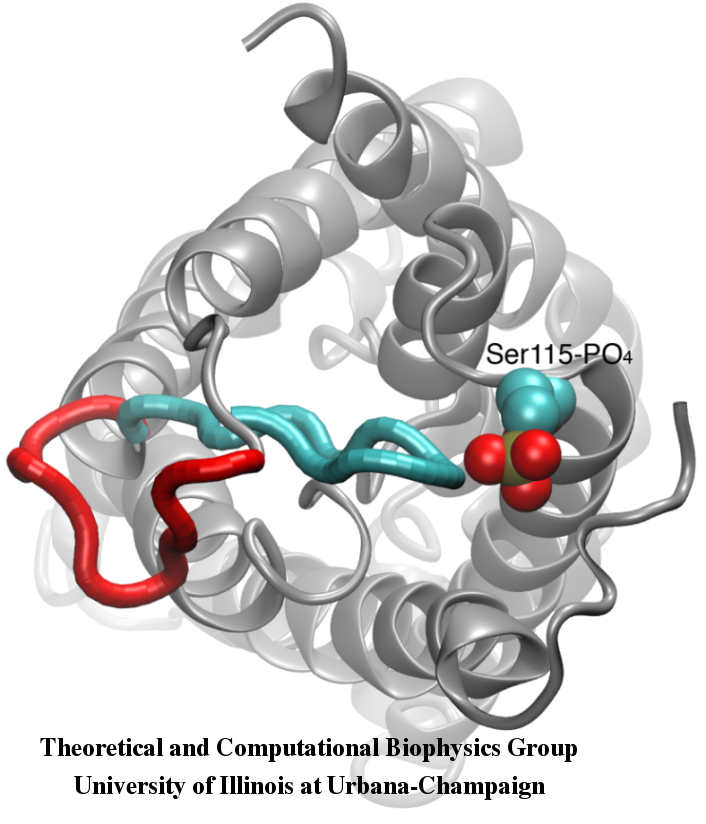
image size:
258.5KB
made with VMD
Your favorite flower pot would not survive a weekend in your office without watering, if it wasn't for a sophisticated cellular mechanism evolved in land plants to conserve water under drought conditions. Water exchange between cells and their environment is facilitated by a group of highly specialized membrane proteins called aquaporins. Although present in all life forms, plants are particularly dependent on their function. While in most species these channels function as always-open "cellular pipes" allowing water in and out of the cell, in plants they evolved into "cellular faucets" whose water permeability can be controlled by the cell. Nearly all plant aquaporins can be gated in response to drought or even flooding conditions, through basic biochemical signals, e.g., phosphorylation and change of pH. A recent Nature paper reporting a collaborative study between crystallographers who succeeded in solving the first structure of a plant aquaporin from spinach, and modelers provides the most detailed view of the mechanism of gating for a membrane channel. Molecular dynamics simulations of the channel performed by NAMD reveals a dual gating mechanism in which phosphorylation of certain protein residues unleashes a long cytoplasmic loop that physically blocks water access to the pore. More information on aquaporin research can be found here.
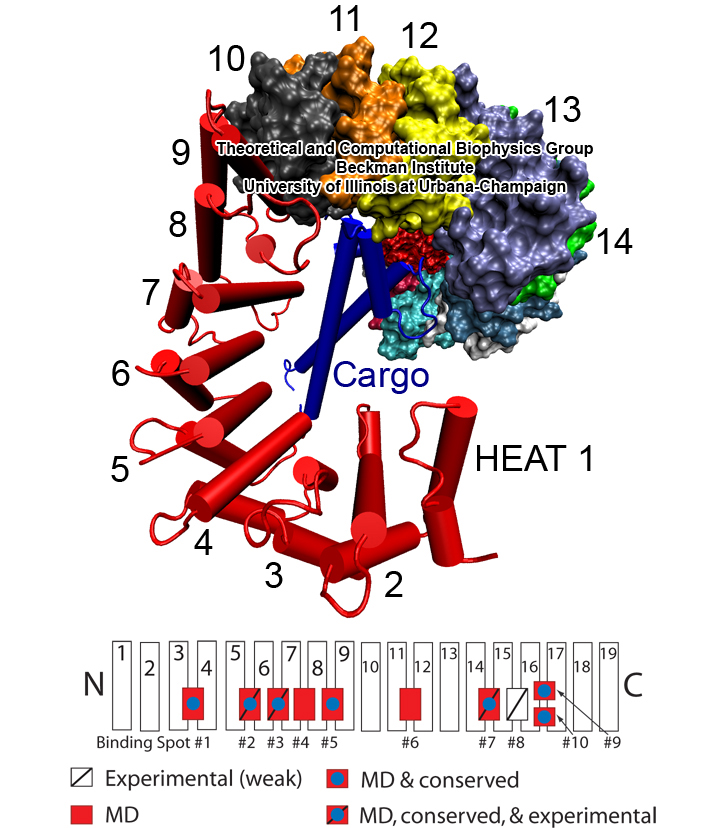
image size:
360.8KB
made with VMD
Eukaryotic cells envelop their genetic material in the cell nucleus whose boundary contains numerous pores. Only small molecules can pass through these nuclear pores unhindered. For all larger ones, passage is highly selective and controlled. The control involves import and export proteins (transport receptors) that load and release cargo on the proper side of the nucleus upon interaction with signaling proteins. Researchers are presently solving the structure of the nuclear pore and its transport receptors with increasing resolution, and the first atomic level investigation into the mechanism of nuclear pore selectivity has recently been reported [paper]. The study inspected the interaction between the transport receptor importin-β with key nuclear pore proteins that appear disordered near the center of the pore and contain characteristic phenylalanine-glycine sequence repeats. Molecular dynamics simulations using NAMD and analyzed using VMD revealed a key insight into the selectivity mechanism. The simulations showed that the key sequences of the repeat proteins interact strongly with certain spots on the surface of importin-β. The study confirmed spots that had previously been identified experimentally and, moreover, found numerous binding spots not yet seen in experiment. Further experiments and simulations promise an understanding of the selectivity of entry and exit from the nucleus, a key element of the cell's genetic control. For more information see our nuclear pore complex webpage.
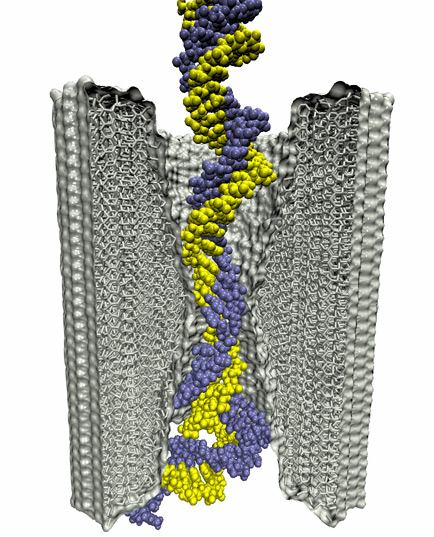
The most celebrated molecule of living cells, DNA, owes its fame to its role as a carrier of genetic information. But DNA is also impressive through other amazing properties, for example its mechanical flexibility. At first sight, it might seem a dull question to ask what is the smallest pore DNA can be squeezed through, as the obvious answer is that the diameter of that pore should be slightly larger than the diameter of a DNA helix. However, recent studies (paper1, paper2) in asking the stated question discovered that double stranded DNA can permeate, without loosing its structural integrity, pores smaller in diameter than a DNA double helix. The discovery was initiated through molecular dynamics simulations, carried out using NAMD and VMD. The simulations demonstrated that if an electrical field, driving negatively charged DNA through a nanopore, exceeds some critical value, the force exerted on DNA stretches DNA to twice its equilibrium length, reducing thereby its diameter and allowing it to squeeze through narrow pores. The simulations predicted precise values of pore radii and associated critical fields. The predictions were validated experimentally by counting the number of DNA copies that passed at different electric fields through synthetic nanopores. Further details about this study can be found here.

image size:
344.8KB
Olga Svinarski and VMD
It was 1995 when NAMD was introduced (Nelson et al.) as a parallel molecular dynamics code enabling interactive simulation by linking to the visualization code VMD. In 1999 a major improvement was accomplished in NAMD 2 (Kalé et al.), scaling to 200 processors at the time due to the efforts and software of the Parallel Programming Lab. NAMD has since matured, adding many features and scaling to thousands of processors, garnering accolades and users in the process. This progress is now collected in a NAMD review paper that presents, in a manner accessible to the novice researcher, the concepts and algorithms behind NAMD, features for steered and interactive MD and for free energy calculation, the elements of the NAMD design that enable parallel scaling, performance comparisons of a variety of platforms, and advice for productive use of NAMD on modern research projects. Case studies ranging from the typical to the elaborate demonstrate the capabilities and flexibility of NAMD. This new reference provides an excellent foundation for working through the extensive NAMD tutorials, either on your own or at a hands-on workshop.
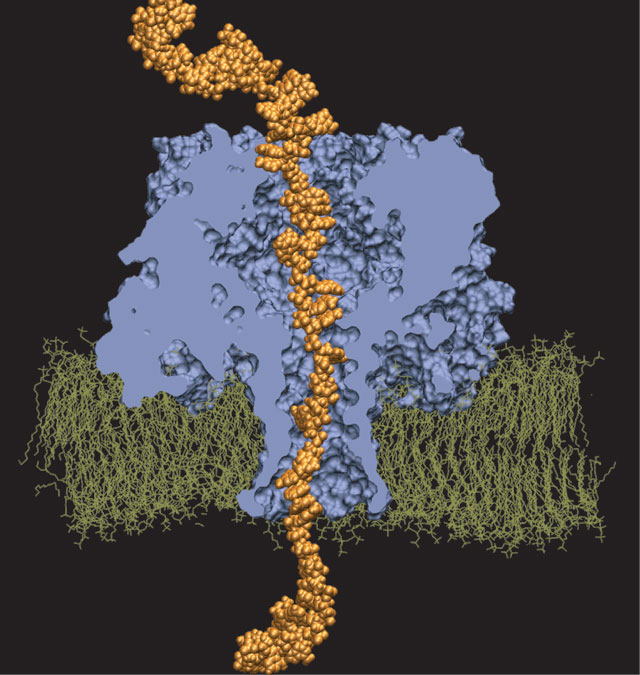
For the sequence of DNA, the genetic instructions of cells, to be read, the double helix of DNA is split open, exposing single DNA strands to DNA binding proteins. Once bound to DNA, the proteins, in carrying out their functions, will crawl along the DNA strand in one of two directions, towards DNA's 3' or 5' end. A recent study of DNA discovered a surprising property of single DNA strands that seems to explain how DNA binding proteins recognize the right direction on DNA strands. By measuring the translocation of DNA through alpha-hemolysin, a membrane protein with a narrow pore, researches discovered that directed single stranded DNA moves much faster when entering the pore 3' end first, rather than 5' end first. The underlying mechanism of this directionality was discovered through molecular dynamics simulations using NAMD and VMD. The simulations revealed that, in a narrow pore, DNA bases tilt collectively towards the 5' end, transforming a wide space directionless DNA brush into a tight space DNA ratchet. The 360,000-atom MD simulation did not only reveal how the DNA bases align and move faster in the "smooth" direction, but did also predict how the directional DNA movement can be discerned by means of direction-sensitive ionic currents through the channel blocked by translocating DNA strands. More details about this study can be found here.
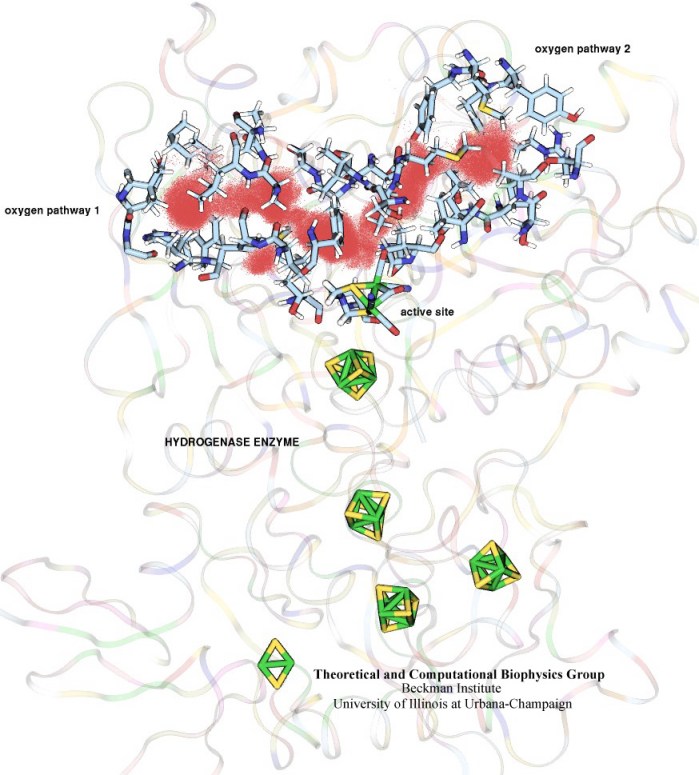
image size:
102.1KB
made with VMD
In an optimistic future, cars and appliances will be powered by renewable energy produced by burning hydrogen gas, with water being the only waste product. To supply this hydrogen gas, scientists are turning their attention to an enzyme called hydrogenase that is found in certain microorganisms, which produce hydrogen gas from sunlight and water. This enzyme, however, is sensitive to oxygen gas, which irreversibly deactivates its hydrogen-producing active site. Understanding how oxygen reaches the active site will provide insight into how hydrogenase's oxygen tolerance can be increased through protein engineering, and in turn make hydrogenase an economical source of hydrogen fuel. In a recent paper (also described in this webpage), the programs NAMD and VMD are used to analyze the gas diffusion process inside hydrogenase, and how it correlates with the protein's internal fluctuations, thereby creating a map of the oxygen pathways. The calculations revealed two distinct pathways for oxygen to reach the active site. Gases participate in physiological processes of many organisms and the new computational strategy developed promises to image gas diffusion pathways for many relevant proteins. In fact, the researchers are currently inspecting hundreds of proteins for their ability to internally transport gas molecules.
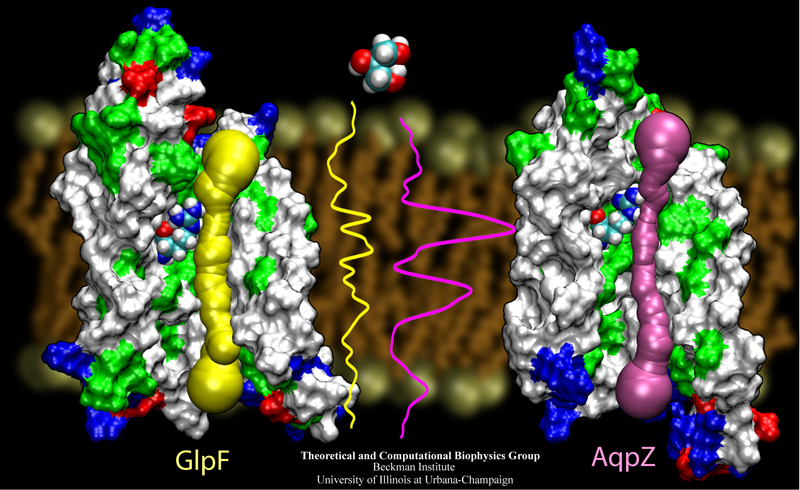
image size:
342.1KB
made with VMD
The import of nutriments over their cellular membranes is one of the main tasks of all living cells. Even though a major part of the cell's molecular machinery is devoted to this task, principles of selective membrane transport are not yet well understood, mainly due to the fact that the membrane proteins responsible are notoriously difficult to resolve in their structure, the latter a prerequisite for a full physical description of the function. Recently, cell biology got very lucky in having the structures of two closely related membrane proteins solved. Two highly homologous aquaporins from the same bacterium, Eschericha coli, have become structurally known: one that conducts only water, called AqpZ, and one that conducts water as well as the nutriment glycerol, called GlpF. The discoveries have permitted us through structure analysis with VMD and molecular modeling with NAMD to look over nature's shoulder in the evolutionary design of two similar import channels of different conductivity. As described in a recent paper and on our aquaporin site, in making a water channel also a glycerol channel, nature has turned to a very basic principle, namely adjusting the overall pore size of the channel from a very narrow channel, just wide enough for water, to one wide enough also for glycerol.

image size: 43.4KB
To guide cell biology research and explain observation through molecular structures and sequence data, life scientists resort increasingly to computational tools. Sequence and structure viewers (VMD) combined with molecular dynamics modeling software (NAMD) are primary methodologies that revolutionized modern biomedicine. The revolution happened so quickly, though, that traditional university training has not kept up with the pace of developments in computational biology. A series of computational biophysics workshops in Perth (Australia), Urbana, Boston, Lake Tahoe, Chicago, and San Francisco attempted to fill the gap through hands-on training. Theory sessions in the morning introduced the concepts and methods used in molecular modeling today; computer laboratories in the afternoon gave participants, students, postdocs, and faculty, opportunities to work through tutorials at their own pace on provided laptops, as well as work on their own research problems. The workshops funded by NIH, NSF, NCSA, UIUC, and UWA met the needs of novices and experts alike for instruction in a new generation of research methods. All workshop materials are available on the web.
image size: 112.7KB
In a biological cell, membrane channels act like miniature valves regulating the flow of ions and other solutes between intracellular compartments and across the cell's boundary. Assembled in complex circuits, they generate, transmit, and amplify signals orchestrating cell function. To investigate how membrane channels work, life scientists, using an extremely fine pipette, isolate a tiny patch of a cell membrane and, in so-called patch clamp measurements, determine electric currents in response to applied electric potentials. Dramatic increase in computational power and its efficient utilization by NAMD allows one today to reproduce such studies computationally, calculating the permeability of a membrane channel to ions and water directly from its atomic structure. In what is one of the largest molecular dynamics simulation to date, described in a recent paper as well as on our web site (here), one copy of the membrane channel alpha-hemolysin, submerged in a lipid membrane and water, was subjected to an external electric field that drove ions and water through the channel. The calculations produced also an image of the electrostatic potential across the channel (see figure).
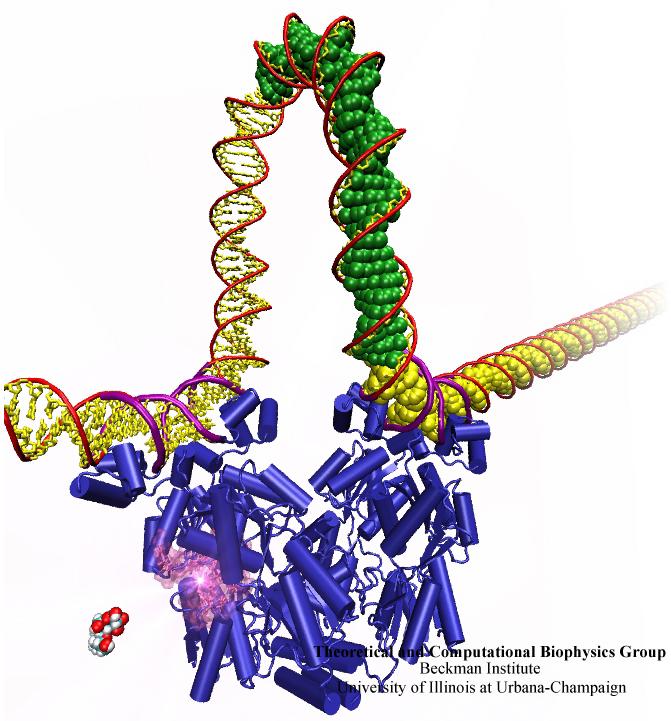
image size:
80.0KB
made with VMD
When Escherichia coli bacteria enjoy lactose and related food molecules in their environment, the cells quickly furnish proteins needed for import and metabolic digestion of the food. A set of genes, called the lac operon, is transcribed into messenger RNA that directs the synthesis of these proteins. When lactose is not available, the protein synthesis would be wasteful and, indeed, is prevented by locking the lac operon. This is achieved by a protein called lac repressor that forces the segment of the lac operon needed to initiate transcription into a loop, but that can be unlocked by a lactose molecule binding to the protein as soon as the food becomes available again. A recent study of the lac repressor combines a 314,000-atom protein simulation using NAMD with a multiscale simulation technique coupling the protein to the DNA loop. The calculations reveal how the lac repressor stretches out two "hands" grabbing the genomic DNA and then keeps a tight grip on the DNA wrestling it into a loop. The discovery is described on our website as well as in a popular article.
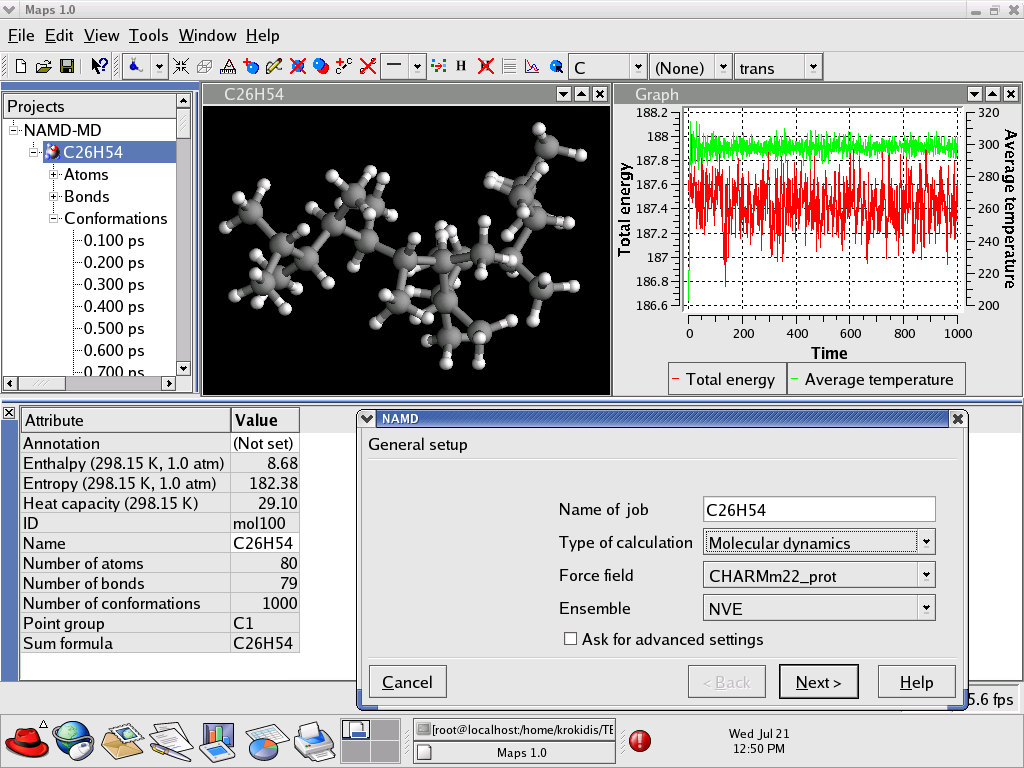
The Materials and Processes Simulations (MAPS) platform from Scienomics is a user friendly environment for molecular modeling and simulations. Its plug-in based architecture enables scientists to use the best technology for a given problem. The MAPS platform runs on Linux, IRIX and Windows® XP operating systems. MAPS includes a series of tools enabling the construction of molecular systems, finite and periodic, 3D visualization and other utilities. The NAMD user can quickly create a molecular model, using standard sketching tools or MAPS' polymers builder, and set up calculations using the NAMD graphical user interface which gives access to many of NAMD advanced capabilities. Analysis tools and graphs available in MAPS enable an easy representation of NAMD results. MAPS' native client-server architecture allows to use the best computational resources available and run NAMD simulations on numerous operating systems. Finally, efficient interaction with office productivity software allows to produce quickly presentations and reports.
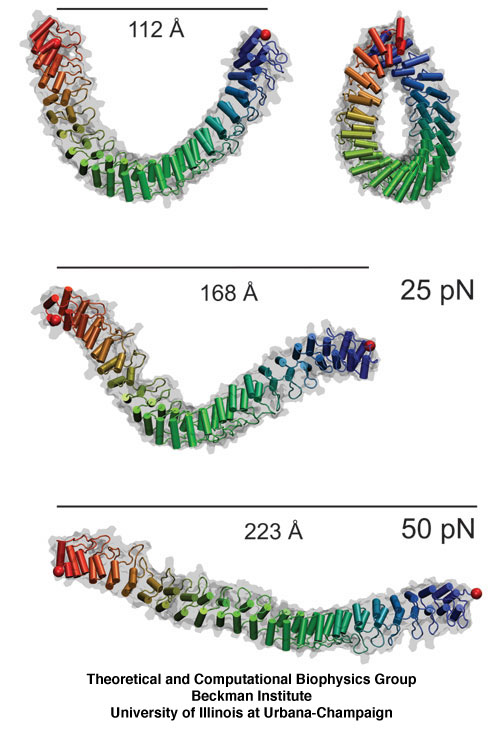
image size:
102.0KB
made with VMD
The ear is a sensitive and robust device, able to perceive the faint sound of flowing water and the thunderous blast of an air plane. Like a microphone, the ear transforms a complex, mechanical stimulus (sound), into an electrical signal, a voltage change in a nerve cell, that can be understood by our brain. This transformation is called "mechanotransduction" and is accomplished by a series of amazingly minute devices that each connect a soft spring to an ion channel, both located in specialized sensory cells, the hair cells of the inner ear. The springs, through their vibrations agitated by particular sound frequencies, control ion currents passing through the channels, thereby, modifying the hair cell internal electrical potential. This leads to neural signaling to the acoustic cortex of the brain. Recently reported molecular dynamics simulations using NAMD, some of the most extensive simulations accomplished to date both in size and duration, showed that the mechanical characteristics of hair cell signaling may be traced to a single protein, ankyrin, that acts as a helical spring. Imagine a soft spring that is extended several inches by the weight of a feather! Ankyrin is such a spring, but a billion times finer (see our ankyrin website).
image size:
138.8KB
made with VMD
Biological evolution left its many traces in the form of organisms as well as in "fine print" in the form of gene sequences and associated protein structures. From the "fine print" researchers can draw conclusions about the inner workings of living cells and derive opportunities to battle disease. Researchers enjoy easy access to sequence and to structure information, but so far mainly separately, i.e., either for sequence or for structure. VMD, our widely used structure viewing and analysis program, has already offered a glimpse of the viewed protein's sequence, but with its latest release has taken a key step further, assisting in viewing and aligning multiple structures and sequences with few mouse clicks. Users of VMD 1.8.3 find themselves routinely comparing their protein of interest with analogous ones getting VMD to color the protein by similarity in structure, in sequence, and showing conserved amino acids. VMD 1.8.3 surprises with numerous further features, including a new cartoon representation that follows the actual molecular structure closely and offers superb, publication quality images. VMD continues to work together with the molecular dynamics program NAMD, permitting viewing and analysis of huge trajectory files by supporting 64-bit processors.
image size:
162.2KB
made with VMD
Biological cells, the basic units of life, are organized assemblies of nanodevices. Nanobiotechnology can adapt Nature's solutions for its own purposes, using computational biology to redesign Nature's nanodevices. In the case of Nanodiscs, bioengineers thought to construct the smallest possible environment that mimics the native environment of membrane proteins. Researchers borrowed the amino acid sequence of a naturally occurring class of proteins, lipoproteins, which are involved in the transport of lipids and cholesterol. The lipoprotein found in humans, apolipoprotein A-1, was used to synthetically engineer "belts" that surround a discoidal lipid bilayer, shielding the hydrophobic lipid tail groups from water. As recently reported, molecular dynamics simulations using NAMD showed an atomic level image of the structure of such a nanodevice. The predicted discoidal shape, diameter, and thickness of Nanodiscs simulated were experimentally corroborated through so-called small angle X-ray scattering. For more details see the HDL & Nanodisc website.
image size:
199.4KB
made with VMD
An important means for generating genetic diversity to provide raw material for evolution and maintain genomic stability is sexual reproduction. At the molecular level, the genes of two individuals are mixed through a process called homologous recombination. This process is found also in many simple life forms, even bacteria. At the beginning of recombination, two DNA duplexes, e.g., from mother and father, are aligned next to one another as the result of homology search, i.e., like strands are brought together with like strands. The four single DNA strands, two in each duplex, cross reciprocally two of the strands between the duplexes. The result is a joint molecule that contains DNA crossovers, named Holliday junctions. The Holliday junction is highly polymorphic in moving along two DNA duplexes, exchanging their DNA. Researchers are now investigating the physical mechanism of Holliday junction migration. The polymorphic, dynamic character of this migration makes observations difficult and the researchers resorted to molecular dynamics simulations using NAMD. The results, reported recently, resolved the dynamics of maternal-paternal DNA exchange through Holliday junction transitions in unprecedented detail providing an atomic level view of sexual reproduction. Check a brief review on our website.
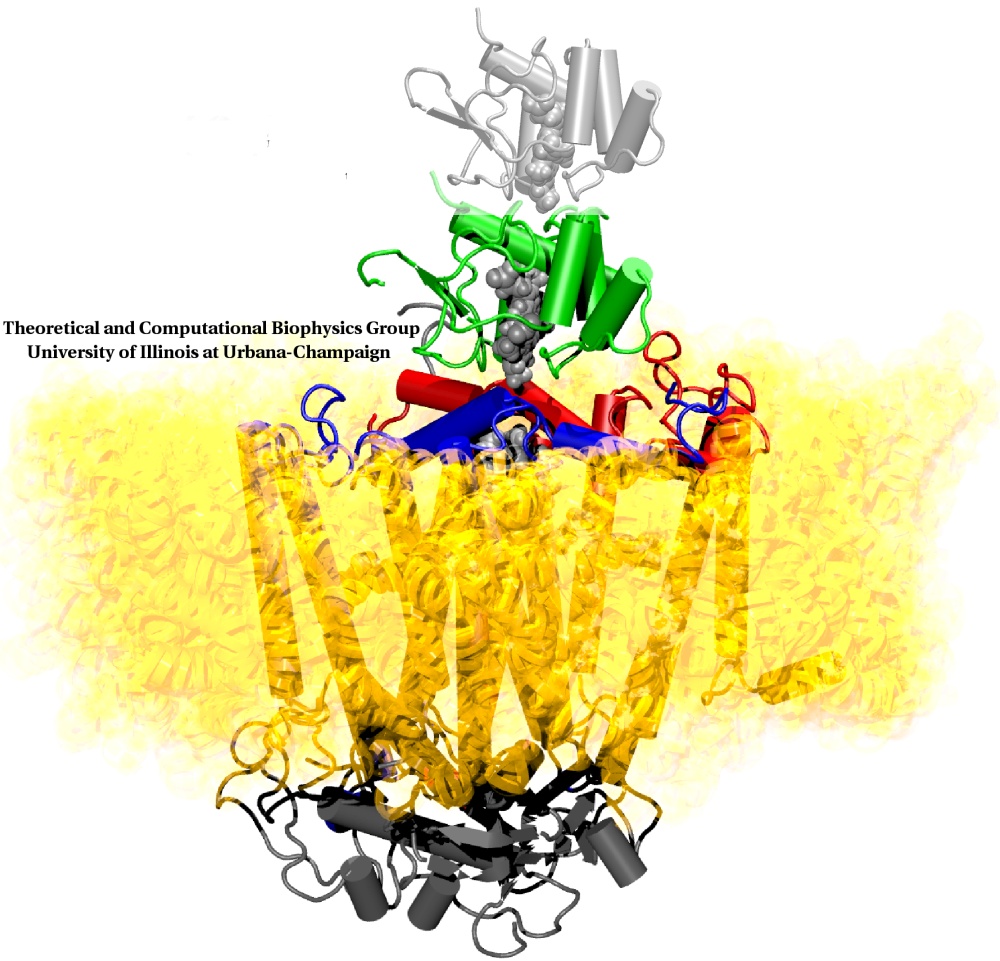
Energy for most of the earth's biosphere is gained when sun light absorbed drives electrons across a membrane through a protein called the photosynthetic reaction center (RC), leaving behind positive electron holes. The electrons join protons to become hydrogen atoms and move, bound pairwise to a quinone molecule, to another protein, the so-called bc1 complex. Here electrons and protons move together back over the membrane and become separated again, thereby establishing an electro-osmotic potential that fuels many cellular processes. However, the electrons need to return to the RC to fill the electron holes left behind. Nature employs for this purpose a kind of bioelectric extension cord in the form of a third protein, cytochrome c2, that shuttles the electrons back from the bc1 complex to the RC. A recent paper reports molecular dynamics simulations using NAMD that investigated how cytochrome c2 plugs into the RC. Landing on a broad face of the RC, interactions steer the protein such that its electron carrying heme group comes close to RC's chlorophylls with electrons missing, a chain of water molecules providing an electrical conduit. The study is yet another example of how simulations provide today complete views of the fundamental processes underlying life.
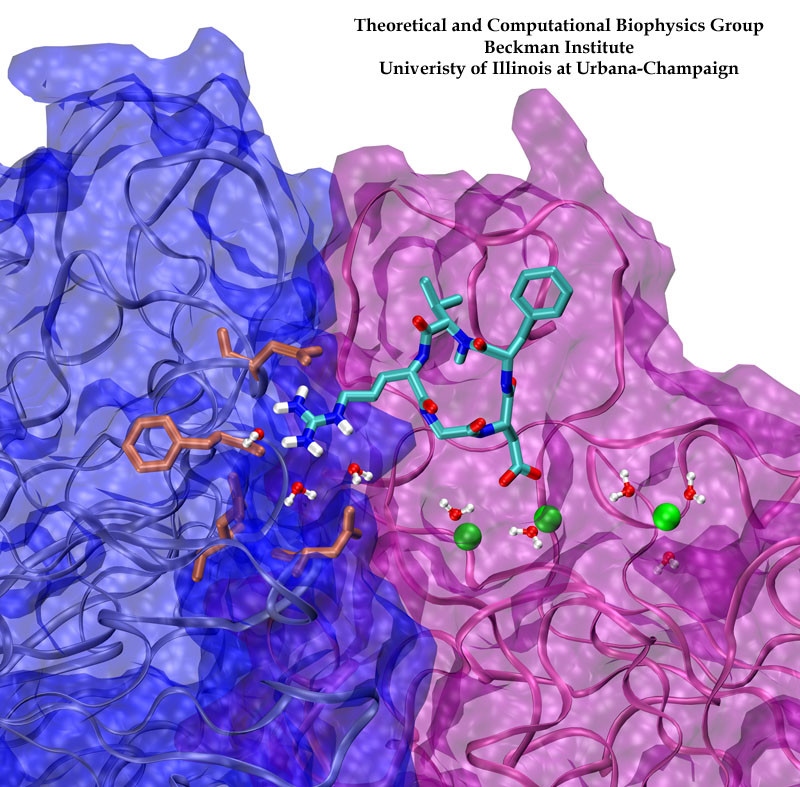
image size:
198.4KB
made with VMD
Biological cells must be capable of attaching themselves to their surroundings. For this purpose they utilize fibrillar proteins, such as fibronectins, that grasp cells through cell surface receptors integrins. The latter act as snap fasteners to the extra-cellular fibrils. The growth, movement, and survival of cells are all dependent on the ability of integrins to fasten cells upon intra-cellular signals or to signal inwards that something has become fastened on the cell surface. The major fastener on integrins are simple divalent ions like Mg++ or Ca++ that can adhere to specific molecules with amazing strength, even though the interaction at the cell surface is exposed to water. Computer simulations using NAMD, reported recently, revealed a dynamic picture of the interactions used by cells to link themselves to the extra-cellular matrix. They showed that it is actually a brave water molecule that is recruited by integrins as a protective shield for the interaction. The simulations provide for the first time a detailed view of how cell tissues are stabilized through surface ions against mechanical stress.
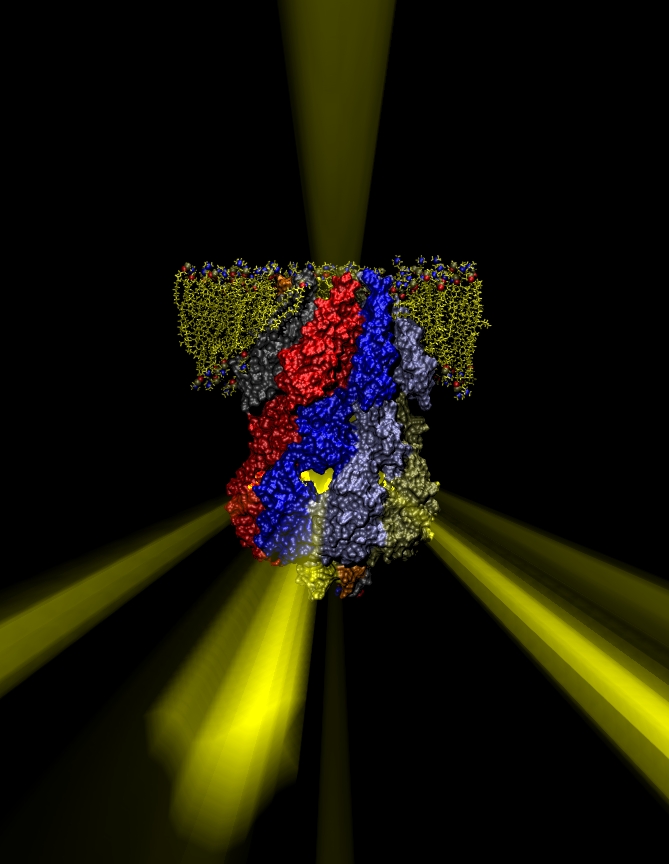
image size:
172.0KB
made with VMD
Many bacteria hang into their cellular membranes proteins that look like a Japanese lantern. These proteins have a flexible cylinder (pore) that crosses the membrane and opens and closes depending on membrane tension; from the cylinder hangs a balloon with seven small openings around its equator (see figure). The apparent function of the Japanese lantern protein, aptly called mechanosensitive channel of small conductance (MscS), is to protect the bacterial cell against osmotic stress: when a bacterium finds itself suddenly in an aqueous environment entering water can burst the cell. Before this happens the cell membrane experiences tension that opens the protein pore, permitting passage of water and ions, the efflux being controlled through the protein balloon. A recent study explores the dynamical properties of MscS, e.g., pore closure and opening as well as ion conduction, by means of molecular dynamics simulations using NAMD. Embedding the large protein into a lipid bilayer and water led to a simulation encompassing 220,000 atoms. Surprisingly, the protein balloon was found to control the arrangement of positive and negative ions through a peculiar pattern of charged, polar, and non-polar amino acids on its internal and external surfaces. This suggests that the Japanese lantern protein has a yet unknown second function in the bacterial cell.
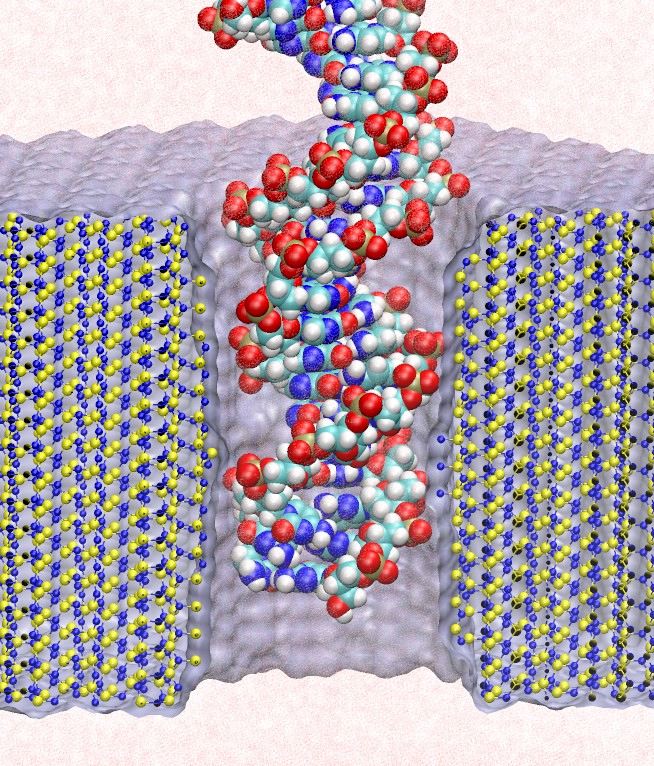
Electrical devices on computer chips built from silicon compounds have reached the small length scales of the building blocks in biomolecules, namely, the amino acids in proteins and the bases in DNA. Using beams of electron microscopes, electrical engineers drill nanometer wide pores into silicon wafers that contain a central layer only a few atoms thick. The engineers surround these pores with transistors and electrodes that can detect charges moving in the nanopore. Electrical fields across such synthetic nanopores can thread charged molecules like DNA through, and electrical signals stemming from single molecules transiting the pores can be recorded. Since the size of the nanopores compares with the dimension of DNA bases, the signals should eventually become precise enough to distinguish DNA bases, such that nanopores can become recording heads reading off sequences of DNA. While such ultrafast recording of DNA sequences is still a distant goal, the manufactured nanopores have been used already for sizing short strands of DNA as reported recently (report1, report2). Molecular dynamics simulations with NAMD and molecular graphics with VMD played a crucial role in imaging the dynamic events (movies available here) involved in recording single molecules of DNA and for optimizing the design of nanopores towards efficient threading and accurate recording. The landmark collaboration between computational biologists and device engineers promises to further unlock the great potential of biomedical nanotechnology.
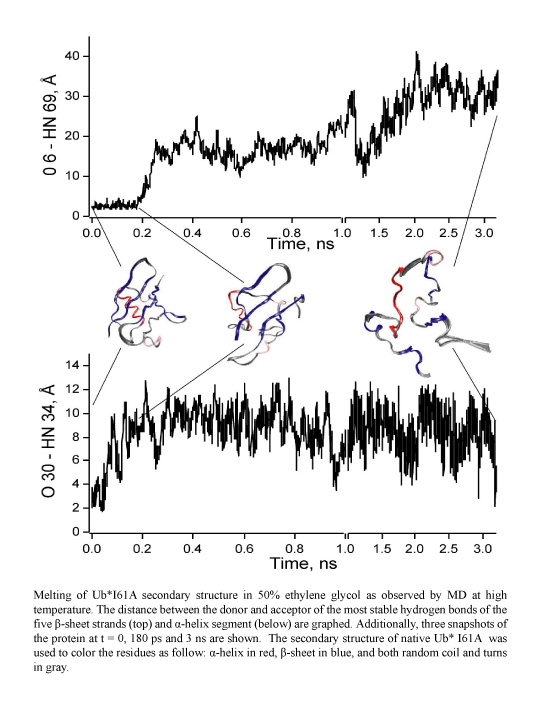
image size:
96.4KB
made with VMD
Proteins perform the many functions of biological cells. This ability arises from the particular three-dimensional structure into which proteins fold at physiological temperature. The quick and precise folding of proteins depends on their molecular environment, e.g., water or lipid membrane, and is being investigated in many laboratories today. A new study from a computational - experimental collaboration investigated the folding and resulting structure of a protein, ubiquitin, in ethylene-glycol, commonly known as antifreeze, mixed with water. In this mixture the folding could be monitored at very low temperature in "slow motion" and resolved in great detail. Computational modeling using NAMD and VMD suggested that adding antifreeze to water leaves ubiquitin folding unaffected and this was born out indeed by further observation. Antifreeze is offering now a wider window into the study of the amazing abilities of other proteins.
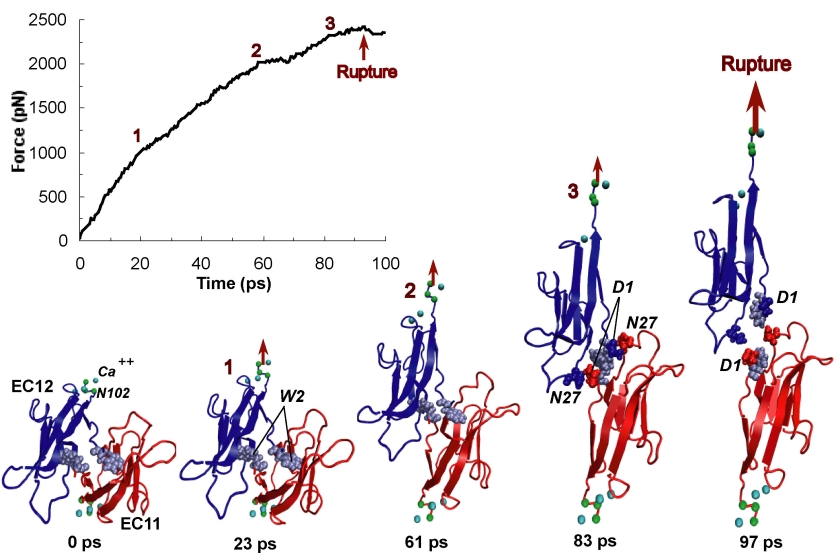
image size:
170.9KB
made with VMD
One of life's great achievements is the development and maintenance of multi-cellular organisms, from an embryo to adulthood. Multitudes of cells need to be sorted and resorted into tissues, organs, and body of living beings. One strategy towards this end is to endow cells with so-called adhesion proteins that connect a mechanical framework inside cells through the cell membrane with other cells. A key type of adhesion protein is cadherin (calcium-dependent adherent protein) that stretches through the cell surface five-tandem domains. The outermost domain can stick to a cadherin molecule from an adjacent cell. Crystallography provided the molecular structures of cadherin pairs and resolved in atomic detail the cadherin-cadherin contact between cells. This prompted a collaboration that aimed at probing the adhesion strength of cadherin pairs through steered molecular dynamics simulations stretching the pairs apart. Results of the simulations were reported in a recent publication that employed NAMD as well as VMD. As shown by crystallography, the cadherins each insert a tryptophan residue into the other protein. The link thereby established can be broken only through strong forces that induce a step-wise slippage of the residues first out of their binding pockets and then along the protein surface. This scenario suggests a mechanism for selectivity among cadherins, i.e., why among the various cadherins found on the surfaces of cells some adhere much better to each other than others.

image size: 137.0KB
Since Leuwenhook, microscopic images of living matter have been produced with radiation, from light to X-rays. With the advent of ever more reliable computational methodologies, molecular dynamics has reached the status of a trusted research instrument. This instrument is particularly powerful for imaging nanoscale, i.e., 10-100 nanometer size, systems. This month our group brought three computers on-line that can serve to image nanoscale systems, three 48-processor rack-mount Xeon clusters (pictured) running our MD program NAMD. One such cluster, a nanoscale system of 300,000 atoms imaged over a nanosecond at the most advanced simulation conditions possible today, requires four days of computing. The Clustermatic software from Los Alamos National Laboratory makes each cluster of 48 processors appear to biomedical researchers as a single machine and allows interactive simulations to temporarily displace long-running NAMD jobs. The clusters have been used already for a study of balance and hearing in the human inner ear. These senses are intrinsically mechanical, relying on hair cells to convert vibration to ion channel modulation. Ankyrin, a protein formed by repeats of a 33-amino-acid domain, is thought to act as a molecular spring in mechanotransduction channels. Explaining the mechanism of ankyrin elasticity requires large simulations of 340,000 atoms that apply repeatedly stretching forces to the protein and monitor its response, revealing a mechanical behavior ideally suited for its biological function. The clusters are presently also used to design artificial nanopores for sequencing of DNA.
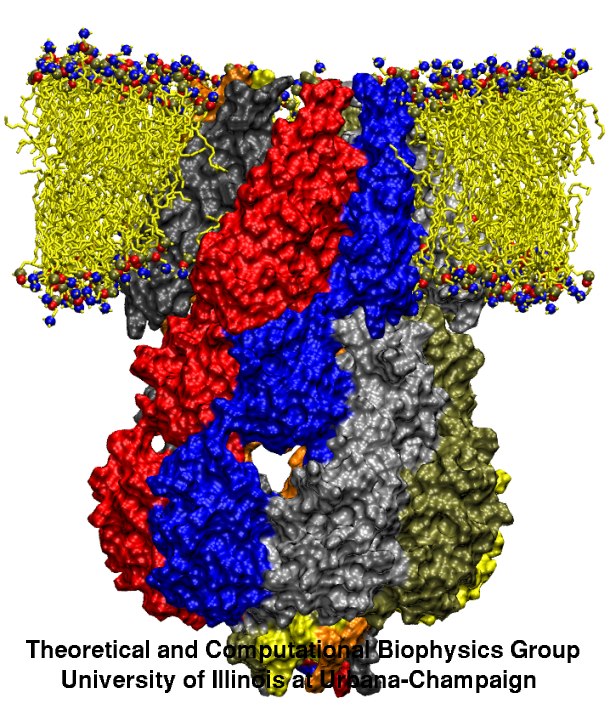
image size:
130.0KB
made with VMD
Computational and experimental biologists investigate jointly the physical mechanisms underlying the function of the molecular machines in cells. Simulations used to encompass biomolecular systems of 10,000 atoms, but recently the size increased tenfold. For example, simulations of aquaporins, a water channel, published during 2001-2003 (1, 2, 3) involved about 100,000 atoms and had been cited in connection with last year's chemistry Nobel prize; simulations of cadherin, a cell adhesion protein, and of ATPase, a key metabolic protein complex, published 2004 included a similar number of atoms. Simulations of over 200,000 atoms for a protein, lac repressor, that regulates genes, and for a protein, MscS, that is a mechanically gated membrane channel will be published later this year. Simulations involving over 300,000 atoms, on a protein, ankyrin, acting as an elastic spring in hearing, have been completed. The increase in the size of simulated systems is prompted by a revolutionary advance in crystallography that resolves ever larger structures of biomolecules and simulations are made possible through the great increase of computer resources at the NSF centers (PSC, NCSA). This marks the beginning of a new era in which systems like virus capsids and the ribosome, entailing 1-3 million atoms, will be studied, too. NAMD (see Highlight Dec 2002) is ready for the challenge posed by simulations needing 250-500 processors today and 1000-10000 processors in the future, to keep up with the developments in the biology laboratories (more).

image size:
130.0KB
made with VMD
Far from playing only the role of bricks and mortar as a mere divider between the inside and outside of cells or between parts of the cell, lipid bilayers are an active, tightly regulated cellular component whose physical properties are critical for the proper function of the membrane proteins contained within them. Lipid bilayers are the preeminent domain of computational biology since despite their considerable stability and impenetrability they form disordered films that are best described through computer modeling, albeit tested by observation. One of the largest molecular modeling projects achieved so far has been recently reported that employed NAMD to investigate the mechanical properties of cellular membranes. The systems simulated were made of lipids and water, composed of about 40,000 atoms, and simulated for over 100 nanoseconds. The simulations revealed that membranes, in terms of their mechanical properties, are far from being homogenous films; rather, they exhibit a delicate multi-lamellar structure of layers that alternatively tend to shrink and expand the membrane, inducing strong forces on all proteins and molecules entering. The lamellar character of the cell's membranes plays a key role for cellular processes such as osmotic regulation and may explain even the action of anaesthetics.
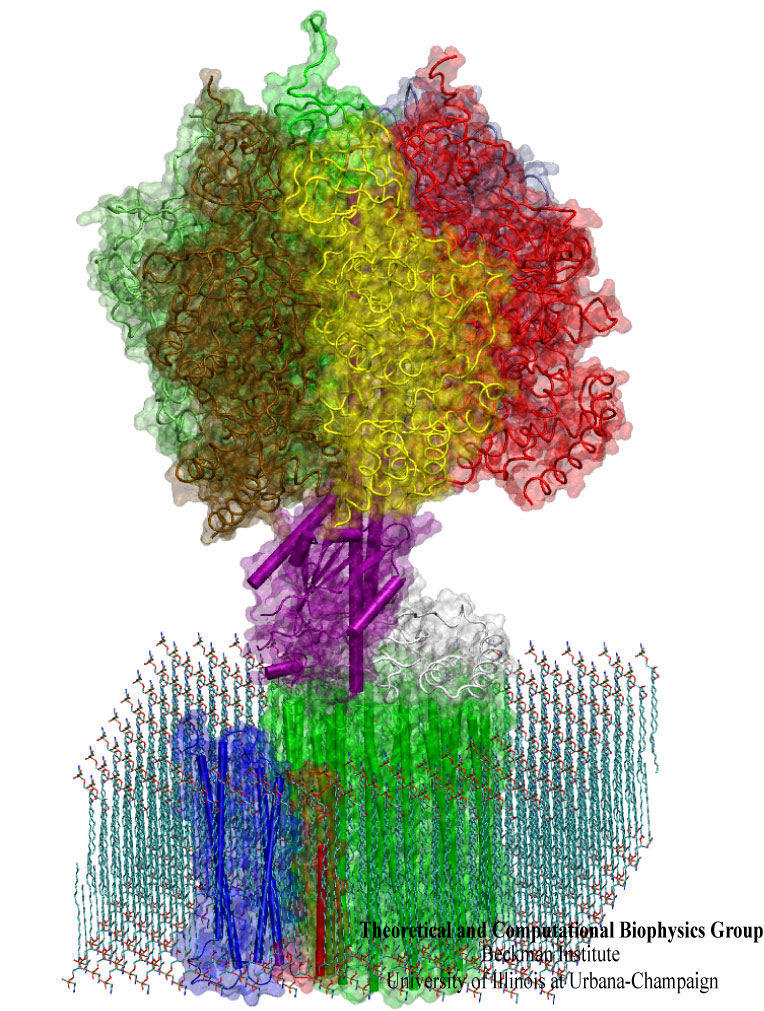
image size:
245.6KB
made with VMD
What will car motors look like in a million years? It's hard to tell, but biological cells seem to have found an ideal engine that they use since the early days of evolution. A spoonful of their engines generates about as much total torque as the strongest car engine today. The engine is called FoF1-ATP synthase and synthesizes the molecule ATP by combining two generator-like motors, Fo-ATPase and F1-ATPase, coupled through a single axle, one motor (Fo) that converts the cell's electrical energy into rotation, another one (F1) that converts rotation into chemical synthesis (see November 2003 highlight). ATP synthase, found throughout the whole kingdom of life, can also work in reverse, turning ATP into electrical energy. Cells typically use the energy of sun light or of food to generate an electrical potential by pumping protons that carry a positive charge across their cell membrane. The energy stored drives the protons back through Fo-ATPase enforcing rotation of the axle; the rotation in turn induces ATP synthesis in F1-ATPase. In one of the largest computational and mathematical biomodeling projects undertaken to this day and reported recently, researchers build from available disjoint structural data a model of Fo-ATPase and demonstrated its function as a motor turning proton conduction into rotation of a cylindrical protein complex. By linking nanosecond molecular modeling to a mathematical model of the motor's key elements, they could follow Fo-ATPase function properly, even when the load arising from driving synthesis in F1-ATPase was added. FoF1-ATP synthase being one of the largest molecular machines in biological cells, modelers needed to employ for its study the most advanced tools, NAMD and massively parallel computers, along with a new approach that combined molecular dynamics and stochastic mathematics.
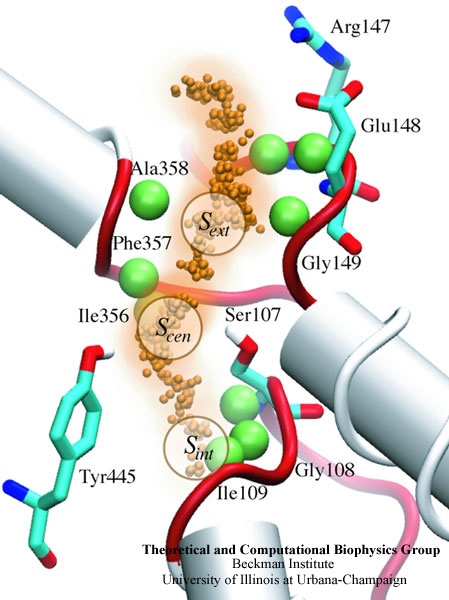
image size:
90.4KB
made with VMD
When their environment gets high in hydrochloric acid, bacteria must protect themselves from chloride ions leaking into the cells. Some bacteria rely on so-called ClC chloride channels to quickly evacuate these ions without letting other small particles pass through. A recent paper investigates the mechanism of chloride transport in a ClC channel from the bacterium E. coli. By means of computer simulations using NAMD, researchers visualized the pathway taken by chloride ions as they pass through the channel and identified how the channel's protein architecture optimizes ion conduction. The relevant architectural features have also been observed in aquaporin water channels (see November 2003 highlight) and the potassium ion channel, but are rarely seen in other proteins. The new discovery demonstrates how much computer simulations are contributing to the emerging picture of life's membrane channel design that is critical for functions of biological cells ranging from the maintenance of our body's hydration to electrical signaling in the brain.
image size:
246.4KB
Movies of stretching FN-III-1:
real media
(
119.5KB
)
windows media
(
125.9KB
)
mpeg (high quality)
(
2.2MB
)
Image and movie made with VMD
Tissues of the human body are composed of specialized cells held together by a connective fabric of proteins, that form the knots of a net glueing cells together. Upon stretching tissues, the knots unravel, rendering the net larger, but mysteriously also firmer. A protein called fibronectin-III-1 plays a particularly important role in the latter respect. Atomic force microscopy revealed that under mechanical tension fibronectin-III-1 stretches to ten times its initial length; but is does so in two steps, the first stretching step leading to net strengthening. It had been discovered earlier that other fibronectins found between cells are made of two sheets packed like a sandwich, but the structure of fibronectin-III-1 remained elusive. In an experimental-computational collaboration reported recently, the structure has now been resolved that at first sight looked similar to the sandwich structure of the other fibronectins, but on closer inspection showed a weak and a strong sheet. Simulations using NAMD revealed that stretching of the protein unravels readily the weak sheet, and only therafter the strong sheet. It turns out that the strong sheet of fibronectin-III-1 by itself, known as anastellin, inhibits tumor growth. Stretching of fibronectin-III-1, as it occurs naturally in tissue, unravels apparently half of the protein to render it extremely adhesive, strengthening a protein net that prevents metastasis of cancer cells and also assists wound healing (press release, more).

image size:
281.7KB
made with VMD
Our "water
permeation movie"
and the Chemistry Nobel Prize
Winner Science illustration competition
This year's Nobel prize in chemistry goes to Peter Agre and Roderick MacKinnon for their groundbreaking work on membrane channels. We join all in congratulating our two colleagues who have advanced knowledge on membrane channels. Foremost, this advance was made possible through great achievements in experimental methodology by several distinguished researchers, but computational and theoretical investigations of the channels the two new Nobelists investigated, contributed significantly. Our group feels fortunate to have participated in the exciting development through modeling studies of the aquaporin channel (see previous highlights June and April 2003, June and May 2002, and November 2001). Our investigations were based on molecular dynamics simulations of channels in their native membrane environment and demonstrate the widening role of computing in modern life science: using the most advanced computers at the NSF centers in Pittsburgh and Urbana, we could establish the mechanisms of conduction and selectivity in aquaporins in a series of studies that are summarized in our aquaporin web page, and in three recent publications in Biophysical Journal that show how the molecular architecture of channels is optimally designed for their function (1, 2, 3).
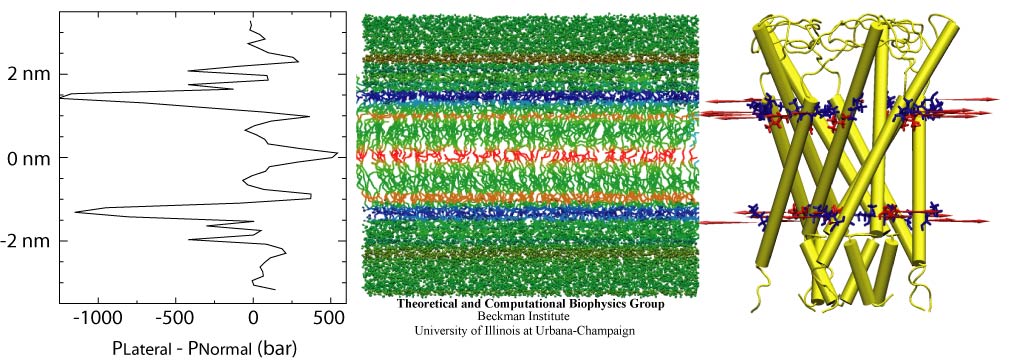
image size: 87.7KB
Living cells sense their environment and respond to its changes through proteins integrated into their outer membrane. These proteins mediate a broad range of cellular activities, including active and passive transport of materials across the membrane as well as response to osmotic shock, which can strain the cell membrane to the point of catastrophic bursting. Cells protect themselves through so-called mechanosensitive channels that open before the membrane tension grows too large. Molecular dynamics simulations and advanced analysis using NAMD and VMD have revealed in a recent report how the joint mechanics of membrane and protein opens a mechanosensitive channel called MscL. The finding promises to revolutionize the modern view of membrane - protein interaction: the membrane, far from being a homogeneous elastic sheet, exhibits a dramatic variation of tension across its thickness that proves to be decisive for the opening of MscL. More here.

image size: 137.8KB
Modeling the molecular processes of biological cells is a craft and an art. Techniques like theoretical and computational skills can be learnt by training, but meaningful applications are achieved only with experience and sensitivity. A summer school in Theoretical and Computational Biophysics attempted to teach both the craft and art of modeling through learning by doing: nearly hundred participants from all over the world came for two weeks to the Beckman Institute in Illinois to stretch proteins, pull water through molecular channels, mine genomic data, build their own computer cluster, and study their favorate biomolecules. After lectures and discussions in the morning, afternoon and evening were devoted to learning by doing, assisted through 300 pages of tutorials, in computer laboratories humming with computational biology software, e.g., VMD, NAMD, and GAMESS, and linked to NCSA's fast pentium machines. The school lasted two weeks, but will go on much longer: all school materials remain available on the web; participants will use BioCoRE to stay in touch and continue the scholarship and friendship experienced in Illinois.

image size:
146.2KB
made with VMD
Biological cells have evolved highly optimized molecular devices made from nanometer size proteins that are the basis for all cellular processes. Engineers seek to conquer the domain of such nanodevices and naturally they look to nature for guidance. In turn, biologists look to nanoengineers for new tools to manipulate cells. A recent study attempts a third type of exchange at the cell - nanotechnology frontier, use of nanodevices that mimic proteins for better understanding of the functions of both: the engineered system can be much simpler than the protein and much easier to comprehend, whereas the protein is more evolved and can reveal the design principles for optimized function. The study links two renowned devices, nanotube and membrane channel. Using the molecular dynamics program NAMD, water flow through nanotubes is simulated and compared with earlier studies of aquaporin water channels in bacteria.
At present, the exchange benefits biology: the very simple nanotube reveals the principles of biologial water conduction, in particular, water flow without loss of electrical voltage. In the future, nanoengineering may benefit, for example, in designing filters for mechanical desalination of sea water.
The import of material into biological cells needs to be highly selective. Which physical interactions are the best capable of differentiating suitable from unsuitable imports? How can one keep import traffic fast despite strong selection? As one knows from border crossing, fast traffic and high security can be conflicting objectives. A recent investigation employed a new simulation method to answer the above questions for import of glycerol through a membrane channel. Linking the renown molecular graphics program VMD with the simulation program NAMD permits researchers to view "live" proteins, e.g., the membrane channel, and employing computer game technology, namely, a device that permits one to pull glycerol through the simulated channel, researchers can feel the mechanical resistance of the pulled glycerol, guide it through the channel, and directly observe the selection process involving glycerol's shape, capability for hydrogen bonding, and electric dipoles. The new methodology, interactive molecular dynamics, will revolutionize computational biology and is one of the first tools that take advantage of fast (TeraGrid) computer networks in linking local graphics workstations (running VMD) with a distant supercomputer (running NAMD). The method has received an enthusiastic editorial comment.

image size:
452.7KB
made with VMD
Infections are battled best by the human immune system when there exists a memory from a previous disease or vaccination. The first step in using this line of defense is recognition: Cells of the immune system capture antigens, e.g., microbes in the respiratory tract, then mature in the lymph system, and finally present on their surface pieces of the antigen to T-cells that may recognize the antigen and become activated. The recognition of the antigen by T-cells is dramatically enhanced through surface receptors, CD2 and CD58, on the T-cell and the antigen presenting cell. The receptors stick out from their cell, adhere to one another, and conjoin the T-cell and antigen presenting cell long enough to enable recognition and activation. The molecular basis of this adhesion has been probed in a recent collaborative study with UIUC chemical engineer D. Leckband. Starting from the available crystallographic structure of the CD2-CD58 complex the researchers carried out 90,000 and 100,000 atom simulations using NAMD and pulled the complex apart in steered molecular dynamics simulations. An analysis of the simulations with VMD revealed in atomic level detail how the human immune system is strengthened through elastic adhesion.
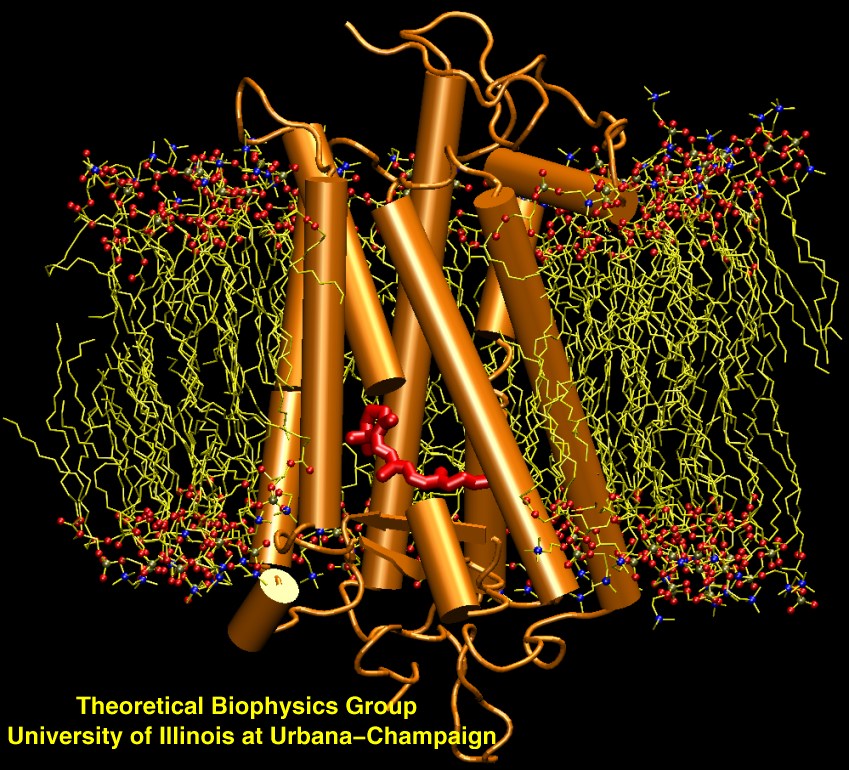
image size:
223.0KB
made with VMD
Scanning their environment for information such as food resources, signs of danger, and illumination is crucial for the well being of biological cells. Evolution has developed for this purpose a great variety of membrane proteins, so-called receptors, that receive physical cues from the external environment through encounters with molecules or absorption of photons and send respective signals into the cell. A common type of receptor sends its signal through interaction with intracellular G-proteins that convey the signal further; proteins of this type are called G protein coupled receptors (GPCRs). GPCRs exist in lower as well as higher life forms and, in fact, the human genome codes for over 1300 GPCRs that detect ions, organic odorants, amines, peptides, proteins, lipids, nucleotides and photons. As about half of modern drugs act on GPCRs, learning how they become activated once they receive their signal is highly relevant. In a recent study, advanced computer simulation techniques using NAMD and VMD have been employed to investigate the first key steps of activation of a GPCR, the visual receptor rhodopsin. more.
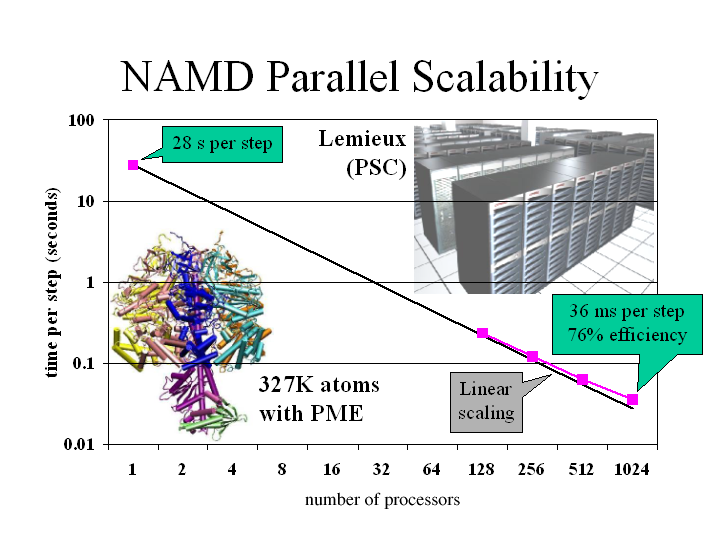
image size: 150.4KB
The parallel molecular dynamics program NAMD, and its sister visualization program VMD, have helped researchers at Illinois discern how muscles stretch, nerves sense pressure, and kidneys filter water. The latter project, for example, used simulations of 106,000 atoms to discover how aquaporins, which are ubiquitous in mammals, plants, and bacteria, allow water to pass while preventing the conduction of protons or ions (article in Science, more). Our years of work developing this software to apply the nation's fastest sup ercomputers to understand the tiny components of living cells were recognized at the SC2002 High Performance Networking and Computing conference with a Gordo n Bell Award for unprecedented parallel performance on a challenging computational problem (pdf of paper-497k). NAMD and VMD are distributed , free of charge, to thousands of scientists in industry and academia around the world, quickening the pace of drug discovery and other vital research to unravel biological processes.
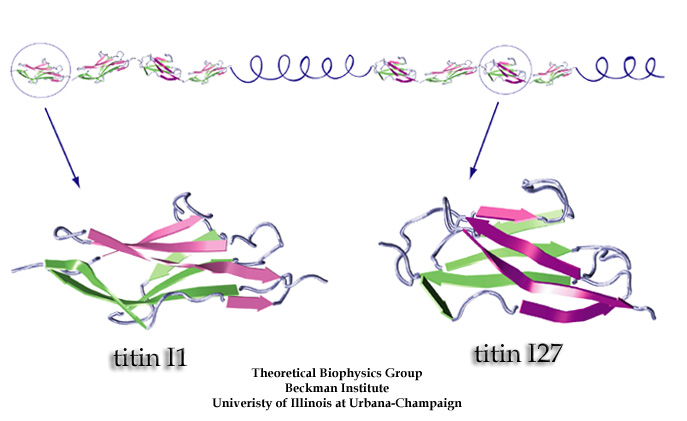
image size:
71.7KB
made with VMD
What kind of function does the longest gene in the human genome code for? The answer is a bit mundane: a very long molecular spring that provides muscle with passive elasticity. Nature adjusts the protein, called titin, for many types of muscle, e.g., skeletal or cardiac muscle, as well as for other cellular functions. The molecular spring contains hundreds of elastic elements in series like beads on a string. One type of bead is the immunoglobulin domain, which can stretch to ten times its normal length. For a long time only one of the immunoglobulin domains was structurally known, permitting only a single peek into nature's design library. Recently, a second domain became structurally known and protein crystallographers and modelers joined forces to discover how nature designs its beads along titin, as described in a recent publication.
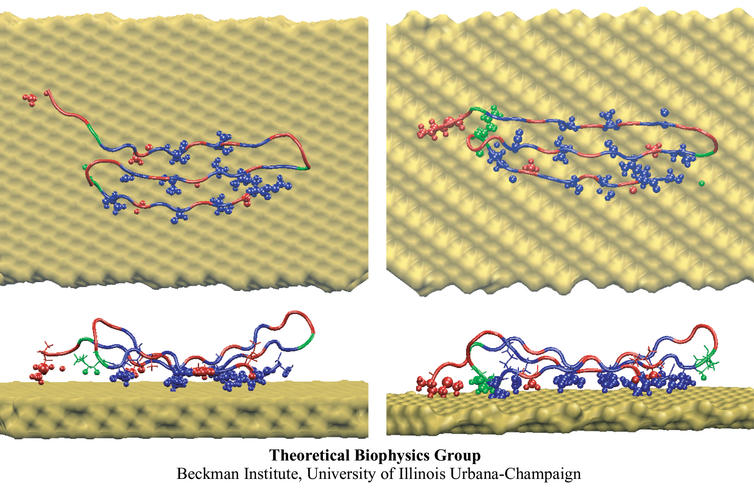
image size:
91.6KB
made with VMD
The biological control of inorganic crystal formation, morphology and assembly is of interest to biologists and biotechnologists studying hard tissue growth and regeneration, as well as to materials scientists using biomimetic approaches for control of inorganic material fabrication and assembly. A molecular-level understanding of the natural mechanisms involved in these processes can be derived from the use of metal surfaces to study surface recognition by proteins together with combinatorial genetics techniques for selection of suitable peptides.
In a recent study, the structure of a genetically engineered gold binding protein has been determined computationally, and the ability of the protein to recognize gold crystal surfaces has been explained. Molecular dynamics simulations were carried out with the program NAMD using the solvated protein at the gold surface to assess the dynamics of the binding process and the effects of surface topography on the specificity of protein binding.
image size:
104.1KB
made with VMD
Living cells rely on nutrients absorbed through their cell membranes, for example on glycerol that is key to the cells' metabolism. Proteins, so-called aquaporins, in the membranes form channels that act as sieves permitting passage of water, glycerol, and like molecules, but prevent other molecules of similar size from entry and clogging. For this purpose the channels interact strongly with molecules attempting to pass. In a recent publication, the energetics of the conduction process of glycerol for the aquaporin GlpF was measured in a molecular dynamics simulation, carried out with NAMD, that pulled glycerol through the channel, monitoring the forces needed to advance along the channel axis. An analysis that discounted the irreversible work done on glycerol, a difficult prerequisite, yielded the energy profile that glycerol experiences along the channel and that reflects how the protein decides which molecules are allowed to pass the sieve.
image size:
82.5KB
made with VMD
Human kidneys need to filter about a bathtub of water a day through cells that contain membrane channels made of proteins called aquaporins. Crystallographers from the University of California at San Francisco (R. Stroud and coworkers) that discovered the structure of one type of aquaporins, aquaglyceroporins, have teamed up with UIUC researchers to determine how these channels achieve their very high water throughput, yet prevent the cells' electrical potential from discharging by not permitting any flow of ions or conduction of protons. The team, combining 106,000 atom simulations using NAMD and crystallography, found that the channels achieve the impressive filtering function by conducting water single file and keeping the water molecules strictly oriented: water molecules enter the channel oxygen atom first and leave the channel oxygen atom last. Aquaporins are ubiquitous in mammals, plants, and bacteria and the finding, published recently in Science magazine, has implications for many biological functions as well as for human diseases, e.g., cataract of the eye, loss of hearing, or diabetes insipidus. (more, press release)
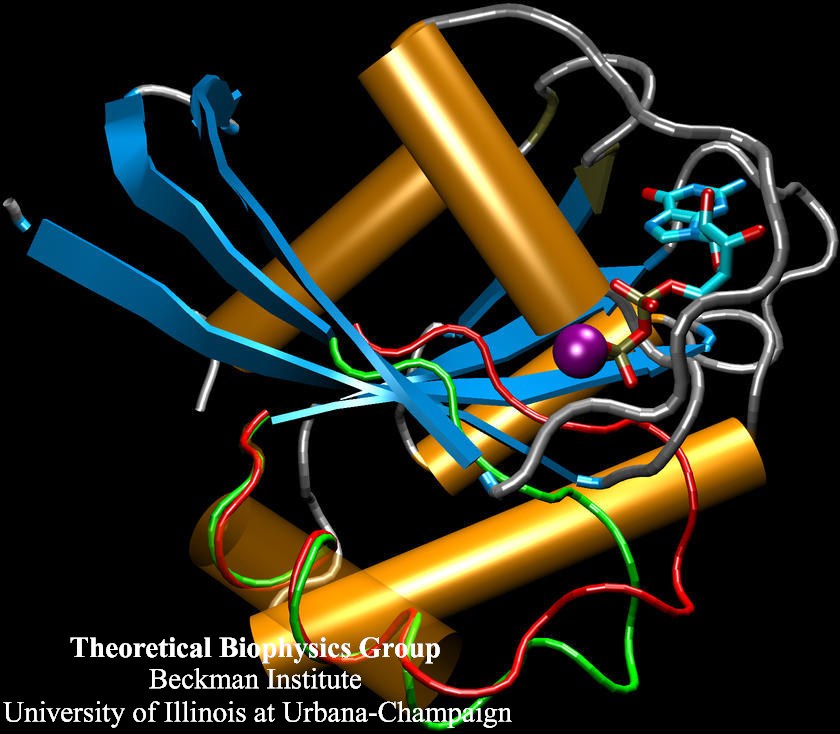
image size:
98.9KB
made with VMD
Biological cells process numerous types of information, for optimal control of their life cycles or to adapt to their environment, and recruit for this purpose signaling proteins. The best known among the latter are the G-proteins, involved in numerous diseases and related to many targets of drugs. G-proteins are closely related to motor proteins: G-proteins get switched on and off through the binding of GTP and its hydrolysis to GDP; motor proteins generate mechanical force through binding of ATP and its hydrolysis to ADP. A recent publication reports a 19,463 atom computer simulation using NAMD that reveals a "power stroke" in G-proteins likewise found in motor proteins. The stroke switches on and off G-proteins' ability to interact with other signaling proteins, with a power stroke that transforms the protein from an ordered into a disordered conformation.
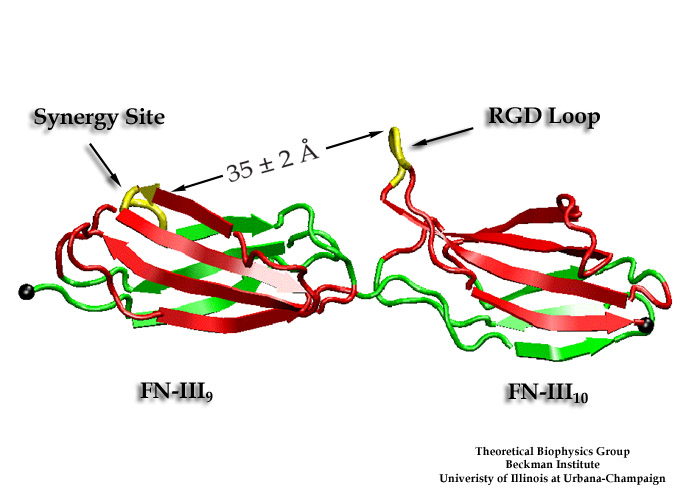
image size:
85.1KB
made with VMD
Cells in animals adhere to dynamic, seemingly random assemblies with other cells that make up tissues like skin, organs, and brain. The cell's adhesion and motion is controlled by the extracellular matrix, with the protein fibronectin being a key component. The proteins have optimal mechanical elasticity and also signal to cell surface receptors, integrins, the tension exerted on them. How this is achieved is the subject of an ongoing collaboration with the research group of Viola Vogel of the Department of Bioengineering at the U. of Washington in Seattle (see also Oct 2001 highlight). The most recent publication from this effort reports a 97,884 atom steered molecular dynamics simulation using NAMD. It is revealed now that stretching two consecutive domains of fibronectin deforms two sites, the so-called RGD and synergy sites as well as their distance. This weakens binding to cell receptors and, as a result, integrins can function as gauges that signal the magnitude of exterior forces to a cell.

image size:
66.6KB
made with VMD
Adenosine triphosphate (ATP) is the fuel of life; every living cell must use ATP to carry out its functions, and the human body synthesizes its own weight in ATP every day. The ubiquitous molecular motor ATP synthase catalyzes the creation of ATP by precisely directing electrochemically generated torque. A detailed understanding of how this system functions can impact areas ranging from neurodegenerative disease research to nanotechnology development. Running at the Pittsburgh Supercomputing Center on LeM ieux, the most powerful computer system in the world for open research, the freely available simulation code NAMD can simulate a solvated all-atom model of ATP synthase with full electrostatics at 65% efficiency on 1000 processors. This achievement in scalability places NAMD an order of magnitude ahead of comparable packages for molecular dynamics simulation.
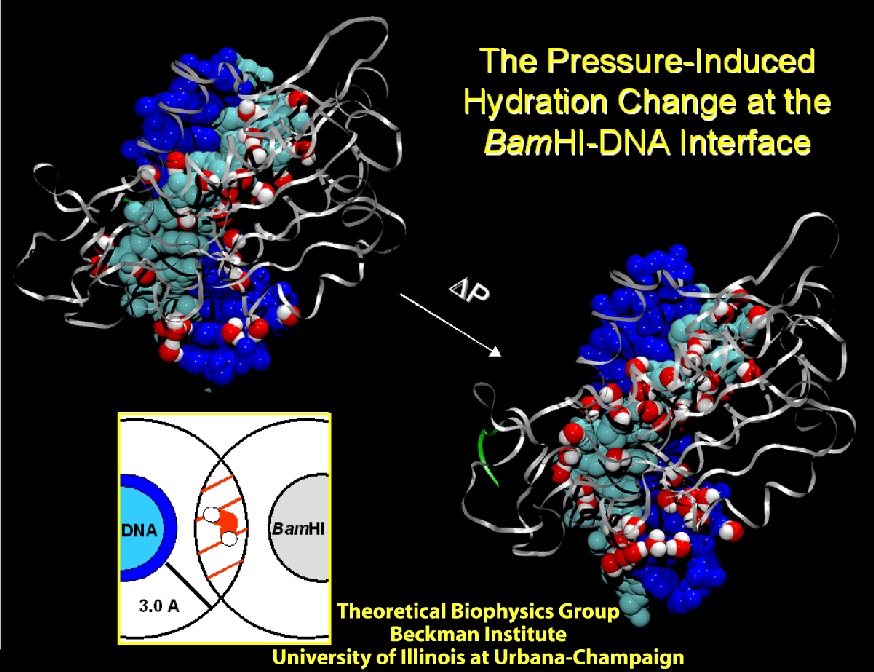
image size:
225.3KB
made with VMD
Deciphering the processes by which proteins recognize and bind to DNA is critical in our quest to understand cellular functions. To reach this goal, a collaboration with the group of Stephen Sligar, UIUC, explored the factors involved in protein-DNA recognition using hydrostatic pressure to perturb the binding of the BamHI endonuclease to cognate DNA. Our joint resulting publication outlines a new technique of high-pressure gel mobility shift analysis to test the effects of elevated hydrostatic pressure on the binding of BamHI (so-called restriction enzyme) to a specific DNA sequence. Upon application of a hydrostatic pressure of 500 bar, recognition between BamHI and the DNA sequence was weakened nearly 10-fold, suggesting an important role of water. An advanced 65,000 atom nanosecond molecular dynamics simulations with NAMD, at both ambient and elevated pressures, complemented the experiments and revealed how water-mediated interactions between BamHI and DNA control sequence recognition.

image size:
246.0KB
made with VMD
Aquaporins are channel proteins abundantly present in all life forms, for example, bacteria, plants, and in the kidneys, the eyes, and the brain of humans. These proteins conduct water and small molecules, but no ions, across the cell walls. Their defective forms are known to cause diseases, e.g., diabetes insipidus, or cataracts. The molecular modeling program, NAMD, along with large parallel computers at the Pittsburgh and Illinois supercomputing centers, permitted researchers now to model aquaporins in the natural environment of membrane and water in one of the largest molecular dynamics simulations ever (over 100,000 atoms). The simulations revealed in unprecedented detail how cells conduct water and glycerol, a molecule that serves cells' metabolism. The simulations provided a movie of the entire conduction process.

image size: 41.0KB
Our low-cost cluster of 32 Athlon PCs (see February 2001 highlight) has been in constant use by local users, providing a substantial and very cost-effective boost to our group's large-scale simulation capabilities. To satisfy demand, we have added two additional 32-processor clusters with higher performance at an even lower cost. On this platform, the freely available simulation code NAMD can complete a 1 nanosecond simulation of the 60,000 atom aquaporin-1 water channel with full electrostatics and constant pressure in a single week. We have given three tutorials, both filled to capacity, introducing participants to cluster hardware and software with the aid of a hands-on session assembling and installing four-processor clusters (see photo).
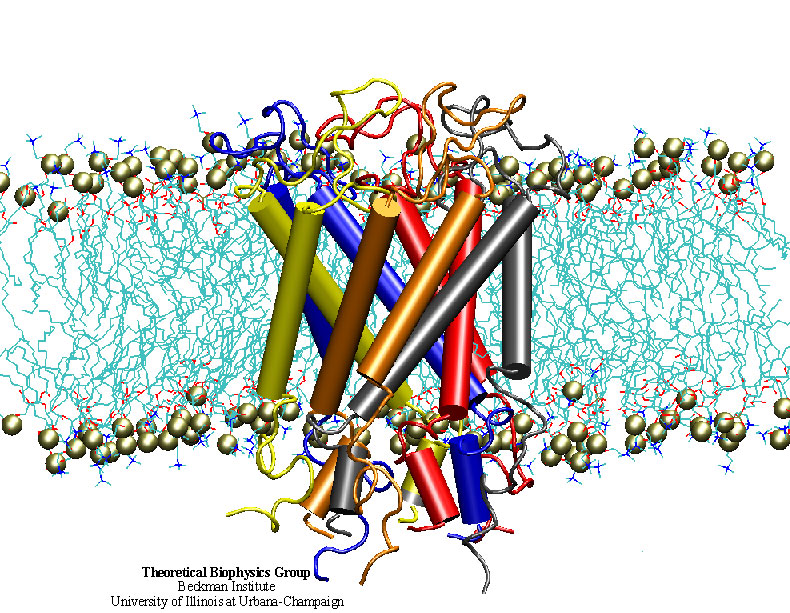
image size:
247.9KB
made with VMD
"How do you feel?" Biologists now have an answer that may surprise you. Our sense of touch relies upon the fact that cells in our fingertips can sense the pressure from a tabletop and transmit a signal to the brain. But until recently, the molecular mechanism for turning the stretching of a cell membrane into a cellular signal was unknown. An important step in understanding this process was the discovery of a protein known as a the mechanosensitive channel of large conductance, or MscL. Though this protein has been studied primarily in bacteria, homologues exist in all major kingdoms of life. Researchers in the Theoretical Biophysics Group have used molecular dynamics simulations to study, at the atomic level, how MscL opens in response to pressure changes. Models of MscL will give us new insight, not only into how we feel, but also how our hearts beat and how we keep our balance. Feel better now? ( more, publication )
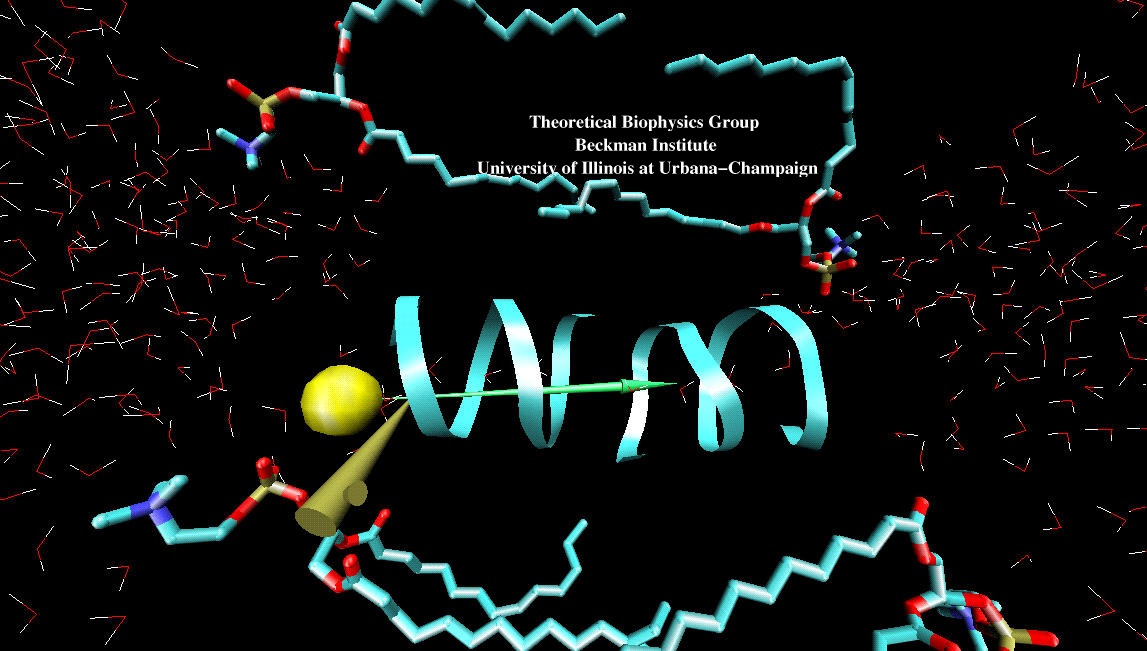
image size:
173.4KB
made with VMD
"If I could just get my hands on that protein!" Single molecule manipulation techniques like atomic force microscopy have brought us closer to this frequently expressed wish. These techniques, however, do not "see" the atomic level detail needed to relate mechanism to protein architectures. True, computational methods do illuminate the elusive protein structures, but are limited to static structures, or trajectories yielded by weeks-long simulations. Now, with the advent of inexpensive, high-performance computing, interactive manipulation of molecular dynamics simulations has become a reality. Linking advanced molecular graphics with ongoing molecular dynamics simulations, and utilizing a haptic device to connect forces from a user's hand with forces in the simulation, researchers can interact with "live" proteins. The new methodology is described in a recent publication and the figure shown here demonstrates a Cl- ion being pulled through a gramicidin A channel (see a 3.8mb Streaming Video).
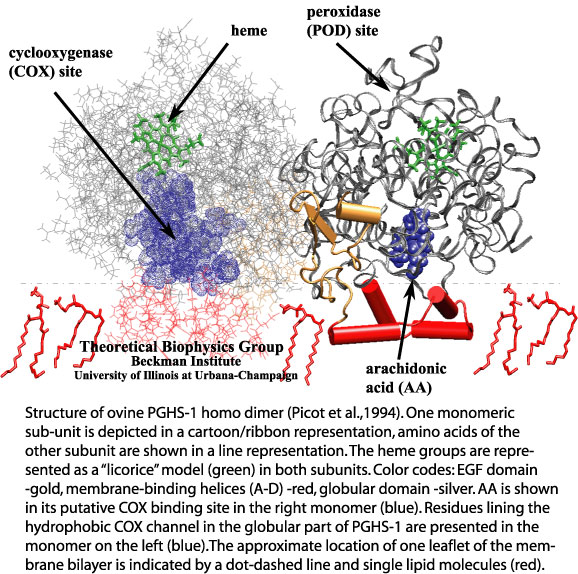
image size:
128.8KB
made with VMD
Aspirin, the widely used pain killer, has revealed many beneficial effects such that it has attracted renewed attention. It has become known that aspirin acts as an inhibitor to prostaglandin synthase. Pharmacological researchers have succeeded to improve aspirin's effect by synthesizing analogue compounds, so-called superaspirins, that target the right type of prostaglandin synthase in the body. The continuing effort has been supported by basic research on the properties of prostaglandin synthases. Molecular dynamics simulations, carried out with our molecular dynamics program NAMD, have investigated how prostaglandin synthases select their substrates, arachidonic acid, through a binding channel that acts as a filter for compounds with the right stereochemical properties. The figure, taken from a recent publication, and made with our graphics program VMD, shows one monomeric subunit (in a cartoon/ribbon representation) of the ovine PGHS-1 homo dimer. To see both subunits click on the image. [More Information]

image size: 25.8KB
We have installed a low-cost cluster of 32 PCs with 1.1GHz Athlon processors, 256MB of RAM, and switched fast ethernet. On this new platform, the freely available simulation code NAMD runs 1 ns of our 92K atom ApoA1 PME benchmark in 8 days with 70% efficiency, the equivalent of a 100 processor Cray T3E. The new machine will be useful for simulations such as the stretching of the muscle protein titin. This work seeks to examine in atomic detail the dynamics and structure-function relationships of this 30,000 amino acid long filament in muscle contraction and elasticity. The cluster also provides a powerful engine for interactive simulations. The Linux-based Scyld Beowulf operating system (see Scyld's press release regarding NAMD) makes the entire cluster appear to computational biologists as a single machine.
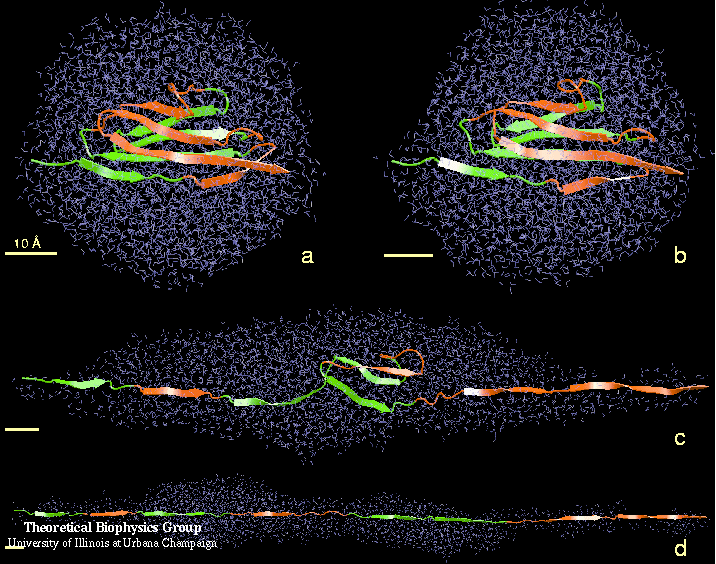
image size:
106.6KB
made with VMD
Water can act as a conformational lubricant for protein folding. The giant muscle protein titin is a roughly 30,000 amino acid long filament which plays a number of important roles in contraction and elasticity. For example, upon stretching in muscle some of titin's protein domains can unfold one-by-one permitting titin to retain elastic properties in muscle over a very wide range of length. To examine in atomic detail the dynamics and structure-function relationships of this behavior, SMD simulations of force-induced titin domain unfolding were performed in close collaboration with atomic force microscopy observations. The simulations led to the discovery that water molecules play an essential role in breaking sets of hydrogen bonds that control the unfolding of titin's domains (see resulting publication).



{kind=link}
{kind=link}
{kind=link}