Bacterial Flagellum




The Flagellum and Bacterial Motility
The most common mechanism used by bacteria to swim through liquid media is the flagellum. The bacterial flagellum consists of 3 major domains: an ion driven motor, which can provide a torque in either direction; the hook, a universal joint which transmits motor torque even if it is curved; and the filament, a very long structure which acts as a propeller, and behaves differently depending on which way the motor turns.
When the bacterial flagellum is rotated by the motor, the filament forms a supercoil, giving it an overall corkscrew-like shape. Flagellated bacteria are able to undergo directed movement through changes in the rotary behavior of the flagellum. When the flagellum rotates clockwise, the filament forms a long pitch supercoil, allowing several flagella on a single cell to form a large bundle, which propels the bacterium along a straight line in a single direction. When the filament is rotated in the opposite direction, it forms a shorter pitch supercoil; this causes the flagellar bundle to disassemble, and the independent motion of several flagella on the cell cause it to tumble randomly. Using these two modes of motion, bacteria can move up or down a stimulus gradient by decreasing their tumbling frequency (if they are moving in the preferred direction) or increasing their tumbling frequency (if they are moving against the desired direction), allowing them to undergo a biased random walk.
Mechanisms of flagellar components
The mechanical properties of all three flagellar components are of interest to biologists (due to the application to pathogenic organisms) and for nanotechnology, since they may offer a template for useful atomic-scale structures. The hook (which acts as a nanoscale universal joint) and the filament (which can be mechanically switched) have been particularly well studied, with both x-ray crystal structures and cryo-EM maps available for both assemblies.
The switching of the supercoils in the flagellar filament is thought to be the result of polymorphic transitions in the filament, when the individual protein units slide against each other. The molecular mechanisms of the polymorphic transition remain poorly understood. It is even unclear which interactions are more important, the protein-protein or the protein-solvent ones. As the resolution in experiments with functioning flagella is not high enough, simulations are necessary to clarify this issue. Likewise, while recent experiments have suggested that the universal joint properties of the hook step from compressibility in the interactions of adjacent subunits along the length of the hook, but the time and length scales required to resolve this interactions are not simultaneously accessible.
Coarse-Grained Model of the Flagellum
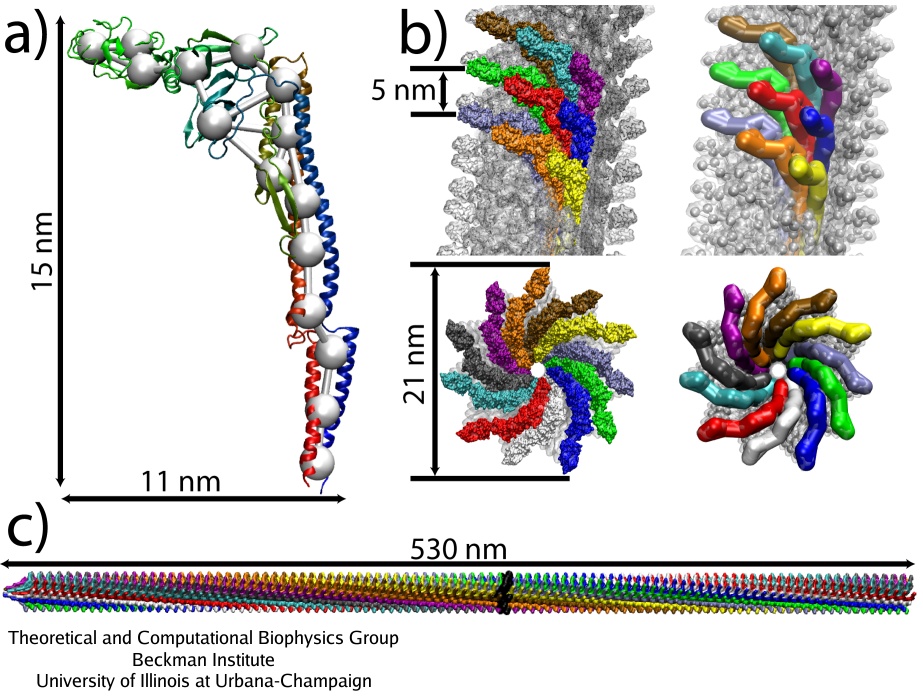 Coarse-grained flagellar filament. (a): Single flagellin monomer, all-atom versus CG model. (b): Arrangement of the monomers in the filament, viewed from the side and from the bottom, all-atom (left) and CG (right). (c): Simulated segment of the filament (1100 monomers).
Coarse-grained flagellar filament. (a): Single flagellin monomer, all-atom versus CG model. (b): Arrangement of the monomers in the filament, viewed from the side and from the bottom, all-atom (left) and CG (right). (c): Simulated segment of the filament (1100 monomers).
All-atom simulations presently cannot reach the time and length scales relevant to the transitions in the flagellar filament (milliseconds and micrometers). Instead, one should take advantage of coarse-grained molecular dynamics (CG MD) techniques. In a CG model, one uses a reduced number of degrees of freedom to describe the system. As a result, with a given computer power, one can simulate larger systems over longer times than what is possible with the all-atom representation.
A CG technique, called the shape-based CG method, has been developed by the TCBG scientists and applied to simulate the flagellar filament (as reported here). In this method, one chooses a number of CG beads that will represent a single protein, and a self-organizing neural networking algorithm is used to distribute the beads so that they optimally represent the shape of the all-atom protein. A single flagellin protein is represented by 15 CG particles (about 500 atoms per CG particle).
The effective potentials for interactions between CG particles were derived from all-atom MD simulations. A simple implicit solvent model, reproducing the dielectric constant and viscosity of water, was used to account for the solvent (the implicit solvent was also parameterized based on all-atom simulations). As a result, a half-micrometer-long segment of the flagellar filament could be simulated over the time scale of tens of nanoseconds. This system consisted of about 20,000 beads in the CG representation, while in the all-atom description it would amount to 70,000,000 atoms, which could not be possibly simulated on modern supercomputers, let alone reaching the microsecond time scales.
The developed CG model distinguishes drastically from the well-established all-atom models. Yet, the TCBG's programs VMD and NAMD could be used without any change to simulate the coarse-grained flagellum. Remarkably, the scaling of the parallel performance that NAMD demonstrated in these CG simulations was the same as normally found in the all-atom simulations. The same CG method was also successfully applied to study the dynamics of viral capsids.
Polymorphic transitions in the rotated flagellar filament
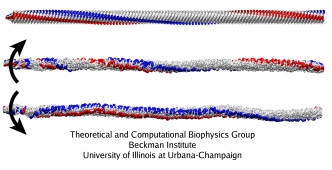 Rotating the flagellar filament.
Rotating the flagellar filament.
Three simulations of a large filament segment (530 nm long, 1,100 subunits) were performed (mpg movie, 6.1M): with the torque applied in the direction corresponding to the running mode, to the tumbling mode, and with no torque applied, as a control. The torque was applied to 30 monomers at the filament's base; each of the three simulations covered 30 microseconds.
Without the torque, the structure of the filament is stable. When the torque is applied, the structure remains stable overall, but the unit proteins rearrange dramatically. As the torque is transmitted along the length of the filaments, parts of the filament start rotating, while other parts (those closer to the tip) are still in rest. In the straight filament, which is the starting structure for these simulations, the protofilaments form a right-handed helix with large helical period. When the torque is applied counterclockwise (as viewed from the base to the tip), the protofilamens remain arranged in right-handed helices, but the pitch of the helices rises; when the torque is applied in the opposite direction, the helices become left-handed. The filament also forms a supercoil as a whole. For the rotation corresponding to the running mode, the filament forms a left-handed supercoil, whereas in the simulation of the tumbling mode, it becomes a right-handed supercoil. The same difference in handedness between these supercoils is found in the living bacteria.
Thus, the simulation reproduces some details of the experimentally known flagellar structures, and also suggests that the rearrangement of the protofilaments upon the rotation is consistent with the previous theoretical models. The protofilaments slide against each other, so that the whole structure undergoes a polymorphic transition from one helical state to another upon the application of a torque. These transitions induce the changes in the supercoiling states of the whole filament, producing the forms that allow a bacterium to swim or tumble, depending on the direction of the torque.
Role of the solvent
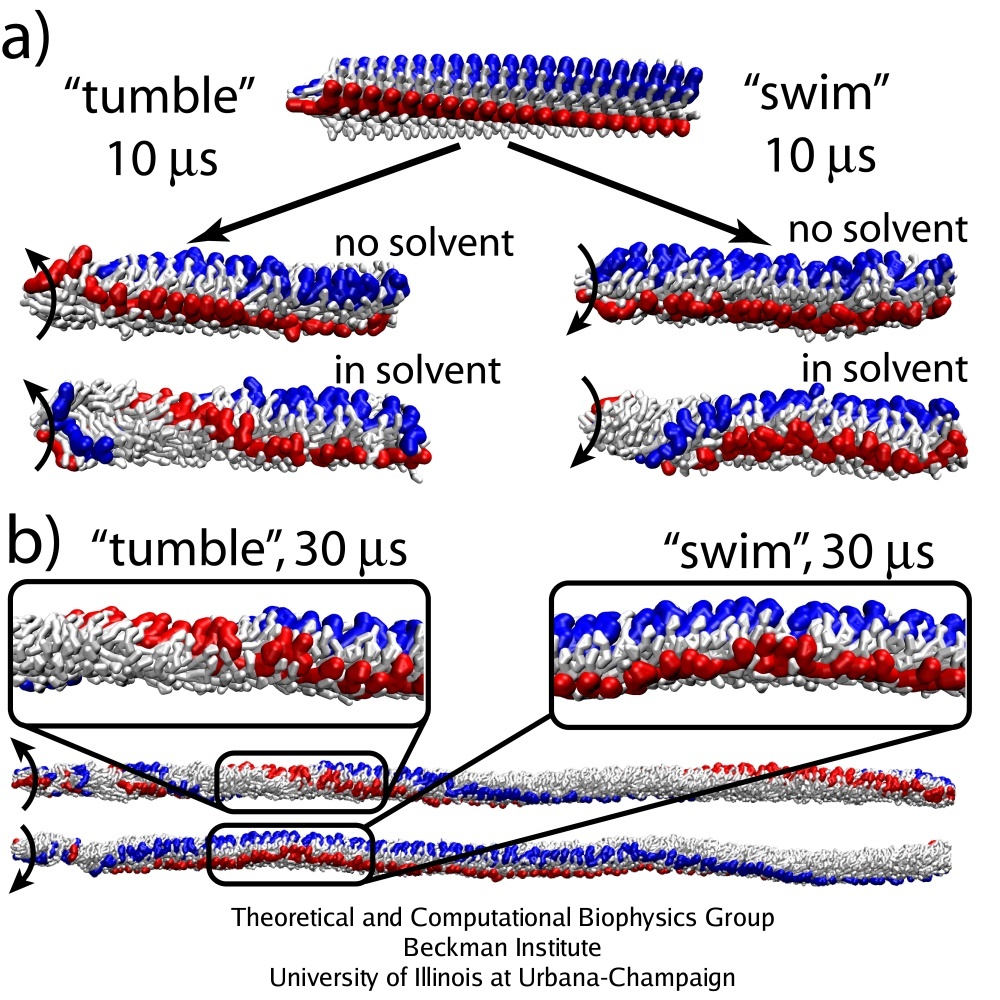 Effect of the solvent.
Effect of the solvent.
Strong coupling between the protein units within a protofilament, as opposed to weaker interactions between the units in neighboring protofilaments, should be the reason why the protofilaments slide against each other so as to produce the observed polymorphic transitions. However, other interactions are also involved in the function of the complex molecular machine that the flagellum represents. Elucidation of the role of certain interactions is often a difficult task for an experiment, but it becomes easy when one takes advantage of numerical computations, where the system can be manipulated at wish of a scientist. The CG simulations of the flagellar filament suggest that the protein-solvent interactions are actually extremely important for the polymorphic transitions to arise, something that has not been taken into account in the most of previous studies of the flagellum.
Rotation of a short flagellar segment (220 monomers) was simulated to investigate the role of the solvent. The speed of rotation propagation, switching of the protofilament states and supercoiling of the segment are the same as in simulations of the 1,100-monomer segment, demonstrating that the observed behaviour is due to local interactions. The short segment was simulated with and without the solvent, which in the employed model means with or without the external viscosity. Without the solvent, the flagellum rotates as a rigid body, i.e., the mutual positions of monomers are frozen, as can be seen in the movies of the rotation for the running mode (mpg movie, 6.0M) and for the tumbling mode (mpg movie, 6.2M). Apparently, the solvent (friction) plays a crucial role in the switching between the arrangements of protofilaments and, consequently, in producing supercoiling along the entire filament.
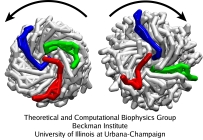
Because the arrangement of the monomers in the flagellum features an intrinsic scoop-like curvature, one wonders if this curvature induces the polymorphic transition due to interactions with the solvent. In simulations where the rotation is applied over the whole length of a short flagellar segment, the monomers brush out for the tumbling mode, and become very smooth for the running mode, with the shape of the monomers being strongly affected by the rotation. Without the solvent (friction), segments look the same for both rotation directions. However, in reality the torque is applied only to the base of the flagellum; in such case, the difference due to rotation one way or another, as observed in the CG simulations, is not in the monomer shape (which is unaffected by the rotation), but in the mutual arrangement of monomers. Thus, the role of the solvent (friction) is not a direct coiling or uncoiling of the filament by rubbing against its ragged surface, but rather a facilitation of proper torque transfer over the filament (by providing friction) allowing the monomers to undergo a polymorphic transition.
Further possibilities.
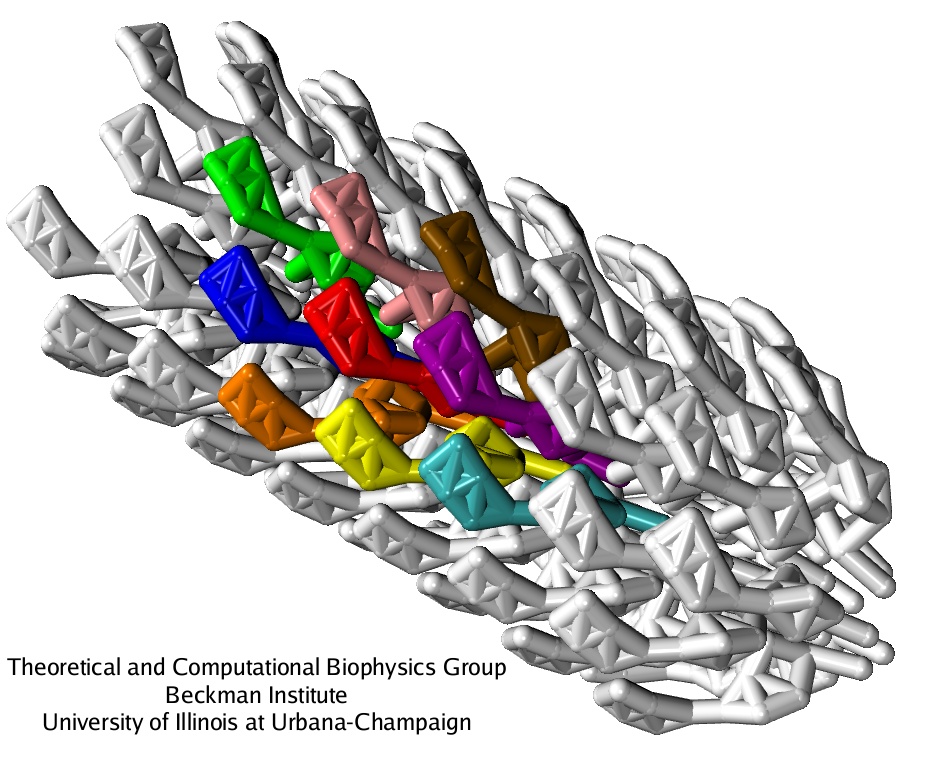
The CG model applied to study the flagellar filament has been deveolped in a general form, and can be in principle used to simulate any macromolecular assembly of known structure. For the flagellum itself, the parts other than the filament should be studied, such as the hook - a universal joint transmitting the torque from the motor to the filament, the connector rings between the hook and the filament, and constituents of the basal body. Eventually, when the structure of all elements is known with the resolution satisfactory fo the CG model (which does not ahve to be an atomic-level resolution), dynamics of the whole flagellum, with its multiple composing prtoeins, can be simulated.
Publications
Investigators
Collaborators
- Keiichi Namba - University of Osaka (Japan)
Related TCB Group Projects
Page created and maintained by Peter Freddolino and Anton Arkhipov.


