
|
Connecting the user-friendly molecular graphics program VMD to the widely adopted MD program NAMD, QwikMD allows its users to setup a molecular simulation in just a few minutes, allowing for quickly study of point mutations, partial deletions or even steering experiments. While making it easy for a new user to perform simulations, QwikMD also works as a learning tool with several "info buttons" where the theoretical background underlying the procedures is provided. |
Recent News and Announcements
VMD 1.9.4 alpha
QwikMD updates in VMD 1.9.4 (alpha). QwikMD had several updates on the latest VMD alpha version and has now the first implementation of the QM/MM interface. In order to run the simulations in the "Live Mode" and/or run QM/MM simulations, the user needs to install the nightly build of NAMD.
VMD 1.9.3 Release
QwikMD is now available on VMD 1.9.3. Employing QwikMD, a user is able to setup MD simulations in just a couple of minutes, allowing quick studies of point mutations, partial deletions and even atomic force microscopy experiments. It is also possible to use QwikMD to prepare Molecular Dynamics Flexible Fitting simulations, which integrates Molecular Dynamics simulations with density maps from cryo-EM experiments.
Availability
QwikMD is available free of charge on VMD 1.9.3 and newer.
To perform Molecular Dynamics simulations you will also need the widely employed NAMD program, which is available free of charge here.
QwikMD beta Linux and QwikMD beta (Windows running VMD 1.9.3) . The latest version of the QwikMD including the most recent bug fixes can be downloaded here (last update on October 30, 2019). Please follow the instructions in the README file included in the qwikmd folder on how to install the beta version.
Employing QwikMD on your research? Please don't forget to cite us: J.V. Ribeiro, R.C. Bernardi, T. Rudack, J.E. Stone, J.C. Phillips, P.L. Freddolino, K. Schulten; QwikMD: Integrative Molecular Dynamics Toolkit for Novices and Experts; Scientific Reports, 6, 26536 (2016)
What is QwikMD?
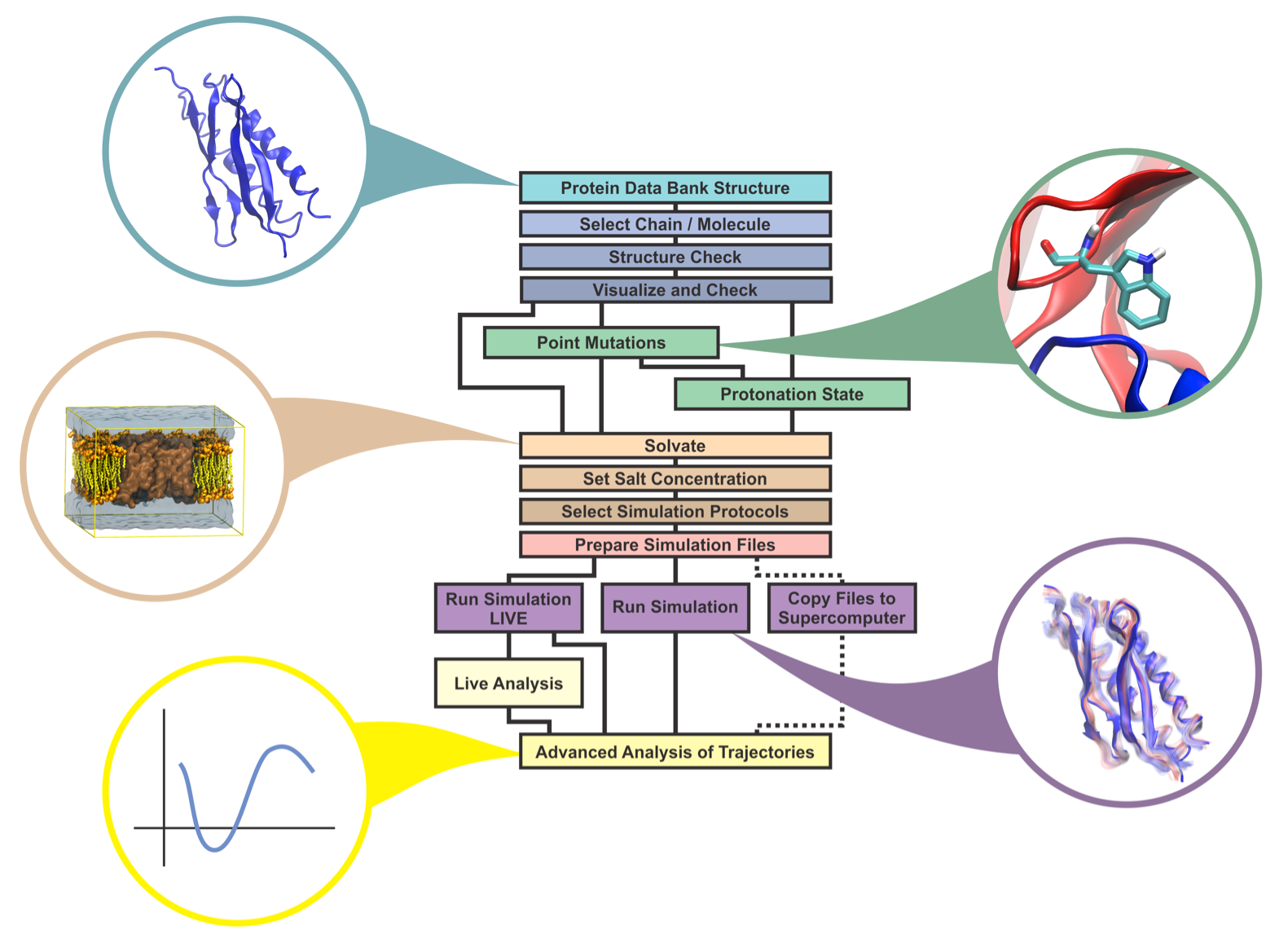
Providing dynamics to structural data from experimental sources, molecular dynamics simulations permit the exploration of biological macromolecules in unique detail. The last advances in molecular dynamics simulations are moving the atomic resolution description of biological molecules to the million-to-billion atom regime, while a variety of sampling techniques are allowing for the exploration of millisecond regime. Although the advances of molecular dynamics simulations are taking us to a new level of knowledge of macromolecular complexes, the vast majority of molecular dynamics users are interested in simple "brute-force" molecular dynamics simulations of relatively simple proteins. In special, structural biology experimentalists are becoming more interested in performing molecular dynamics simulations to improve their understanding of protein structure/function relationship. To help these experimentalists and also beginners to MD to transcend the initial learning curve barrier of any MD simulation software, we developed a new tool that connects VMD to NAMD, two of the most popular simulation programs. Employing our tool, namely QwikMD, a user is able to setup a molecular simulation in just a few minutes, allowing for quickly study point mutations, partial deletions and even steering experiments. While making it easy for a new user to perform simulations, QwikMD also works as a learning tool with several "info buttons" where the theoretical background underlying the procedures is provided.
Incorporating the most widely used features of NAMD and VMD, QwikMD provides a graphical user interface (GUI) for the standard MD workflow (see image below). Built on this workflow, QwikMD automates all necessary steps while checking for common errors and ensuring reproducibility of the result by recording each step performed.
Spotlight: VMD 1.9.3 Brings Simulations Into Focus (March 2017)

image size:
2.2MB
made with VMD
The latest release of VMD brings many advances that help researchers prepare, analyze, and visualize molecular simulations. The new QwikMD plugin streamlines key simulation preparation and analysis tasks, and guides users in the creation of reusable simulation workflows and protocols. VMD now includes several advanced features for parallel analysis and visualization of cellular-scale simulations, as reported here, and here. VMD 1.9.3 strengthens collaboration between experimental and computational biologists by supporting a broader range of experimental density map image formats, such as those used in cryo-electron tomography. Many updated plugins are included in VMD 1.9.3, including tools for analysis of free energy perturbation simulations, MDFF hybrid structure fitting, ffTK force field parameterization, and normal mode analysis. VMD 1.9.3 adds support for new hardware and operating system platforms including IBM OpenPOWER (ORNL Summit), a variety of GPU-accelerated ARM SoCs, the Amazon AWS EC2 cloud, and most recently, the Intel Xeon Phi Knight's Landing many-core CPU (TACC Stampede 2, Argonne Theta). The VMD 1.9.3 release adds stunning graphics produced using interactive ray tracing using the latest multi-core CPUs and GPU accelerators, enabling 360-degree panoramic movie rendering for VR headsets, as reported here, and here. Interactive ray tracing makes the task of getting a molecular image "just right" much easier than ever before; it also enables rendering of spectacular movies for communication of scientific results. A VR movie rendering tutorial assists users with the steps required in rendering and encoding VR movies for upload to YouTube for display using VR headsets such as Google Cardboard, Oculus Rift, and GearVR. More details about VMD 1.9.3 features can be found here.
Support & Bug Report
The first version of QwikMD is available in VMD 1.9.3.
Attention, this is the first public release of QwikMD. Even though the software was exhaustively tested, QwikMD might still have some bugs so use caution.
If you find any problems using QwikMD please contact us by using our BUG REPORT form, or by e-mailing one of QwikMD's main developers: jribeiro@ks.uiuc.edu, rcbernardi@ks.uiuc.edu, or trudack@ks.uiuc.edu.
Any feedback is appreciated.
QwikMD Main Features
Easy Setup of MD Simulations
Point Mutations
Changes in Protonation
Protocols for MD, SMD, MDFF
Live View Simulations
Integrated Basic Analysis
Info Buttons to Guide Novices
Advanced Protocols for Experts
Setup Membrane Environment
Advanced Analysis
Full log Capabilities
Easy Reproducibility
Available on Amazon Cloud
Training & Workshops
QwikMD is an important part of our training workshops. You can check our upcoming workshops where your can learn more about Computational Biophysics here.
Documentation
Development
QwikMD is in constant development by a team of investigators in Professor Klaus Schulten group at University of Illinois. The QwikMD main developers are:
How to use QwikMD?
For those who are experts in both Molecular Dynamics and Computational Biophysics, using QwikMD should be intuitive. For beginners, QwikMD is a perfect tool that helps overcoming the initial learning barrier imposed by most Molecular Dynamics software. The "info buttons" present in QwikMD help both beginners and experts to take the best advantage of Molecular Dynamics simulations. Each "info button" generates a window where the user can find fundamental information related to Molecular Dynamics simulations, such as the description of solvent models and protein structure, as well as short explanations of the options available in QwikMD graphical interface and the actions triggered by the buttons. For more information, the user is redirected to web-pages through links present in the bottom of the information window.
Please check out our one page tutorials, which are useful for a quick start. A complete tutorial is available on our tutorials website.
Download one page tutorials:
- QwikMD - How to perform Glycoside Hydrolases simulations.
- QwikMD - How to perform simulations of Peptide + Membrane.
- QwikMD - How to perform steered molecular dynamics simulations of ultrastable cellulosome components.
- QwikMD / MDFF GUI - How to perform MDFF of Adenylate Kinase.
QwikMD / MDFF GUI tutorial files:
Scientific Successes
QwikMD is the newest software developed by our group and it was officially released in the Fall of 2016. However, QwikMD was already employed in some breakthrough research:
Oridonin Targets Multiple Drug-Resistant Tumor Cells as Determined by in Silico and in Vitro Analyses, Kadioglu et al., Frontiers in Pharmacology, 2018
Molecular mechanism of extreme mechanostability in a pathogen adhesin, Milles et al., Science, 2018
NAMD goes quantum: an integrative suite for hybrid simulations, Melo et al., Nature Methods, 2018
In silico-based identification of human α-enolase inhibitors to block cancer cell growth metabolically, Lung et al., Drug Design, Development and Therapy, 2017
Combining in Vitro and in Silico Single-Molecule Force Spectroscopy to Characterize and Tune Cellulosomal Scaffoldin Mechanics, Verdorfer et al., JACS, 2017
Skeletal Dysplasia Mutations Effect on Human Filamins' Structure and Mechanosensing, Seppälä et al., Scientific Reports, 2017
Structural insights into the functional cycle of the ATPase module of the 26S proteasome, Wehmer et al., Proceedings of the National Academy of Sciences, 2017
Structure of the human 26S proteasome at a resolution of 3.9 Å, Schweitzer et al., Proceedings of the National Academy of Sciences, 2016
Making hybrid QM/MM simulations easy
Biomolecular force fields fail to address chemical reactions and other complex quantum effects near, e.g., reaction centers. Treating such parts of the molecule via quantum mechanics (QM), while the majority of simulation remains classical, allows the accurate representation of quantum effects in relatively large systems, such as enzymes. While many levels of quantum theory are available, QM/MM simulation requires the exchange of only positions, charges, and forces, enabling a common NAMD interface serving multiple QM engines.
In NAMD, a QM/MM interface to both ORCA and MOPAC has already been developed and is available in the nightly build of NAMD. QwikMD fully supports QM/MM simulations in VMD 1.9.4 (alpha) version.
Read more about QwikMD
1. QwikMD: Integrative Molecular Dynamics Toolkit for Novices and Experts; J.V. Ribeiro, R.C. Bernardi, T. Rudack, J.E. Stone, J.C. Phillips, P.L. Freddolino, K. Schulten; Scientific Reports, 6, 26536110 (2016)
2. NAMD goes quantum: an integrative suite for hybrid simulations; M.C.R.Melo, R.C. Bernardi, T. Rudack, M. Scheurer, C. Riplinger, J.C. Phillips, J.D.C. Maia, G. B. Rocha, J.V. Ribeiro, J.E. Stone, F. Neese, K. Schulten, Z. Luthey-Schulten; Nature Methods, (2018)
3. Easy and Fast Setup of Molecular Dynamics Simulations: Combining VMD and NAMD for Experimentalists; J.V. Ribeiro, R.C. Bernardi, T. Rudack, K. Schulten; Biophysical Journal, 110(3), 641a (2016)
4. Making Classical and Hybrid (QM/MM) Molecular Dynamics Easy and Fast with QwikMD; J.V. Ribeiro, R.C. Bernardi, T. Rudack, K. Schulten; Biophysical Journal, 112(3), 448a (2017)
5. QwikMD-Gateway for Easy Simulation with VMD and NAMD; J.V. Ribeiro, R.C. Bernardi, T. Rudack, K. Schulten, E. Tajkhorshid; Biophysical Journal, 114(3), 673a-647a (2018)


