Protein Folding
The ability of proteins to fold into their native state is essential for cell function; misfolded proteins not only lose their function, but can also cause neurodegenerative diseases, including Alzheimer and Huntington. Study of protein folding can aid in preventing protein misfolding diseases and in designing proteins with novel functions. Although most cellular proteins fold on timescales of milliseconds to seconds, several small proteins have been designed and characterized experimentally to fold on a timescale of less than 20 µs. The project will employ Center-developed long-time MD simulation with NAMD to investigate: (1) the folding pathway of λ-repressor and (2) the effect of mutations on folding of the λ-repressor.
Spotlight: Protein Pressure Cooker (Apr 2014)
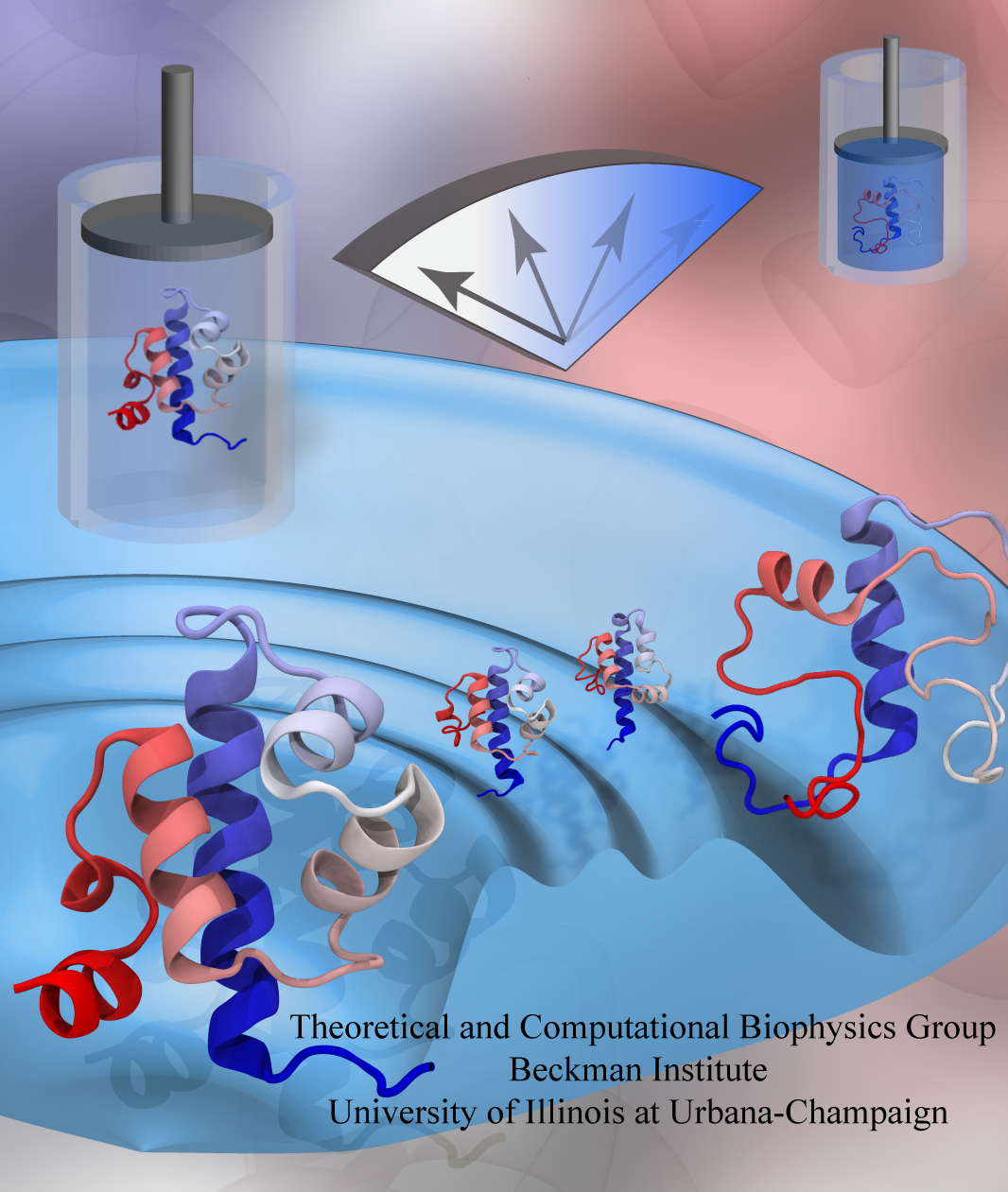
While pressure can help in cooking your favorite meat for dinner, pressure is also helping scientists to study how proteins, a key ingredient in any meal, loose and regain their proper shape. Proteins are key building blocks for any life form on earth, making the many machines that drive living cells. For any protein to do its job correctly, it has to first settle into the proper shape, the so-called native state. The process, referred to as protein folding, is still a mystery (see July 2012, Nov 2009 and May 2008 highlights). A general consensus is that the mystery can be solved only through a combination of experimental observation and computer simulation. In two recent reports (1 and 2), a team of experimental and computational scientists have used high pressure to force a protein to loose its proper shape, similar to what happens in a pressure cooker. After the high pressure is released in the experiment, the protein regains its proper shape, apparently by following two folding pathways, one fast and one slow. Using the molecular dynamics simulation program NAMD, as well as a special purpose supercomputer, Anton, the researchers were able to identify these two pathways and to follow every single folding step of the protein at an unprecedented precision. The studies greatly improve scientific understanding at the molecular-level of how proteins respond to pressure changes and, while not giving delicious recipes for pressure cooking in the kitchen, they are likely to suggest how to use pressure to dissolve toxic proteins that arise in disease, such as in Alzheimer's disease. More on our protein folding website.


