MD Simulation of Protein Folding
Topics: Overview - WW domain - Villin Headpiece - λ-Repressor - PublicationsOverview
Proteins, made up of specified sequences of amino acid building blocks, are the workhorses of biological systems, performing most of the tasks necessary to maintain life. One of the greatest challenges in molecular biology today is that of determining how the sequence of a protein -- the exact ordering of amino acids it is composed of -- specifies its structure and function. Protein folding mechanisms have been extensively studied over the past few decades through both experimental and computational means, although the long timescales required for folding processes have meant that simulation of complete folding trajectories in explicit solvent were not possible until very recently. Instead, protein folding simulations have generally made use of coarse models, implicit solvent, or the use of very large ensembles of shorter trajectories to obtain information on the physical folding pathway.
Recent advances, however, have made combined experimental and computational studies of protein folding possible through the development of proteins that fold on the microsecond and even sub-microsecond timescale, and through advances in molecular dynamics (MD) simulations allowing simulation of multiple microsecond folding trajectories within a few months on modern supercomputers. Our ongoing simulations on protein folding will attempt to directly link all-atom folding simulations with folding kinetics data from the Gruebele lab at UIUC. Through simulations of a variety of protein mutants with different folding rates, we hope to gain a general understanding of factors driving protein folding.
Pin1 WW Domain
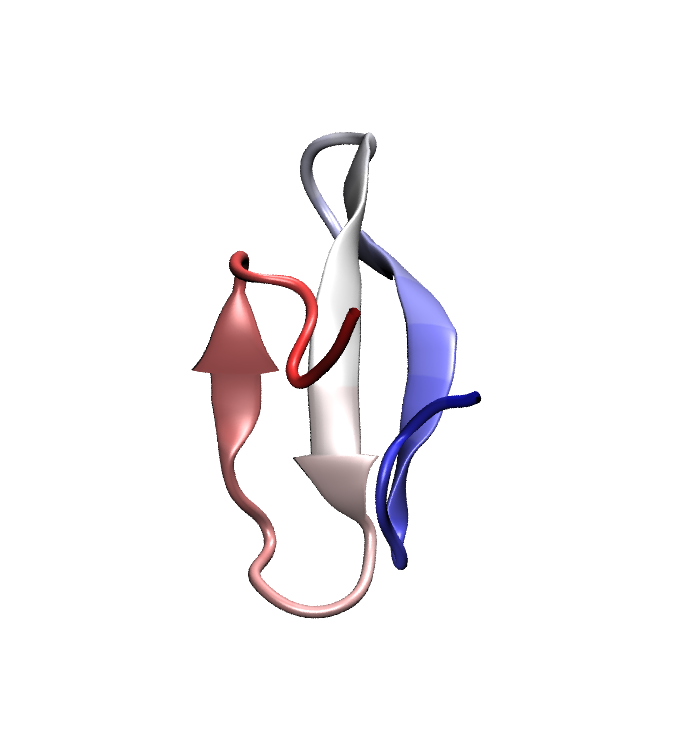 Folded conformation of the Pin1 WW domain. Click to view a folding trajectory.
Folded conformation of the Pin1 WW domain. Click to view a folding trajectory.
The WW domain is a fast folding, three strand beta sheet domain present in a wide variety of proteins. The WW domain from human Pin1, a proline isomerase involved in controlling cell proliferation, has been chosen as an exemplar since a number of fast-folding mutants of this protein exist and have been characterized through temperature-jump experiments. Even the fastest folding WW domain mutants require a few (3-5) microseconds to fold, which is at the very edge of the timescales currently accessible to modern all-atom molecular dynamics. Using a specially tuned version of NAMD, a 10 microsecond simulation of Pin1 WW domain was recently obtained starting from a fully unfolded state; this effort marks one of the longest single MD trajectories ever obtained, to our knowledge. Unfortunately, in this simulation the protein misfolded into an alpha helical state which remained stable throughout most of the trajectory.
Based on a single simulation, it is impossible to determine whether the misfolding that we observed was due to a single aberrant trajectory, systematically slower dynamics in simulations compared to the real system, or thermodynamic stabilization of the helical, misfolded states. To aid in distinguishing between these possibilities, we performed three additional 4 microsecond simulations; in all three cases, a set of helical conformations formed that was similar to those observed in the original 10 microsecond trajectory. The additional simulations make it extremely unlikely that the failure of the WW domain to fold in MD simulations was simply due to a few unlucky simulations, but still do not distinguish between kinetic and thermodynamic reasons for the prevalence of helical states. Temperature-jump experiments in the Gruebele lab on a newly designed mutant which should destabilize any helical character present in the WW domain failed to accelerate folding (and, indeed, slowed it down), which make it less likely (but not impossible) that the alpha helical conformations are physically realistic.
Conformational free energy calculations
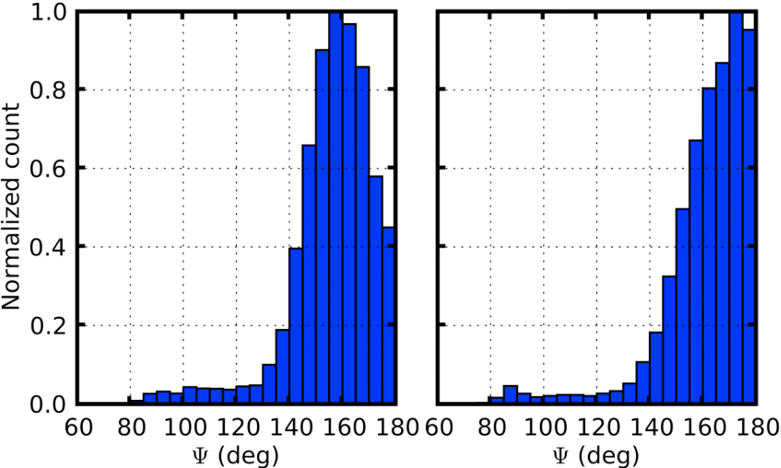 Distribution of backbone hydrogen bonding angles from WW domain simulations of helical (left) and sheet conformations.
Distribution of backbone hydrogen bonding angles from WW domain simulations of helical (left) and sheet conformations.
In order to definitively determine whether the WW domain misfolding was due to kinetic or thermodynamic problems, free energy calculations were carried out using the newly developed Deactivated Morphing method of our collaborators Sanghyun Park and Benoit Roux. Free energy calculations in biological simulations generally face the challenge that one must identify some path that can be followed between the conformations of interest, which can be very difficult for major structural rearrangements of proteins. Deactivated morphing solves this challenge by following an "imaginary" path in which the internal interactions of the protein are removed, so that it can then be morphed to a target structure. From deactivated morphing calculations on the WW domain, we found that the helical states were indeed thermodynamically favored by the force field that we used, suggesting the need for future efforts to refine the helix/sheet balance in MD force fields. One key contributor to the relative instability of the sheet conformation was the pattern of backbone hydrogen bonding, which are incorrectly oriented due to the lack of directionality in hydrogen bonding in modern MD force fields (see figure at left). While quantum mechanical calculations and a survey of the PDB indicate that the angle about the hydrogen should have a distribution centered between 155 and 160 degrees (see, e.g., Kortemme et al., J. mol. Biol. 326:1239 (2003)), in MD simulations the sheet conformation of the WW domain has a distribution that is much closer to linear, introducing energetic frustration that may destabilize the native sheet structure relative to the helical structures.
Villin Headpiece
One of the best studied examples of fast-folding proteins,
wild type villin headpiece is known to fold in 4-5 microseconds;
in addition, a fast-folding mutant exists which folds in under a microsecond.
The villin headpiece has been the target of a wide variety of
experimental and computational efforts to characterize its folding;
however, at present, no atomic-scale predictions regarding the
folding mechanism of villin headpiece have survived experimental
scrutiny, and thus the details of the folding of this apparently
simple model system remain unknown. Part of the challenge in
computationally studying villin folding is undoubtedly a matter
of resources; even for this fairly small system, until recently no
full-length folding trajectories had been obtained.
 Folded conformation of the villin headpiece. Click to view a folding trajectory.
Folded conformation of the villin headpiece. Click to view a folding trajectory.
We have performed a series of MD simulations of villin headpiece folding in explicit solvent in order to study the folding mechanism of villin and understand how folding is accelerated in the fast-folding mutant.
In three separate trajectories (movies: 1, 2,
3) wild type villin was found to
fold after 5-8 microseconds; these trajectories represent the
first all-atom, explicit solvent MD simulations of villin folding
on realistic timescales. The early stages of folding were very
different between the trajectories, exploring a variety of different non-native conformations in each case. Near the end, however, all
trajectories come to a common path: all of the secondary structure
elements of the protein form, but arrive at a conformation where
one of the helices is flipped
relative to the rest of the protein (key steps in the transition are shown below). Folding can only occur after
the helices fully dissociate from each other and then come back
together in correct (i.e., folded) orientations. The results
of an example trajectory, shown at right, illustrate folding
to a native state in 5.5 microseconds. The consistent folding
path followed by the villin trajectories late in folding agrees
with experimental findings that a single rate-limiting transition
dominates the folding of the protein, and provides information on
the nature of this transition that is impossible to obtain through
other means. Based on the simulations we were able to identify a
set of mutations on the flipped helix that would destabilize the
trapped folding intermediate and thus are expected to accelerate folding.
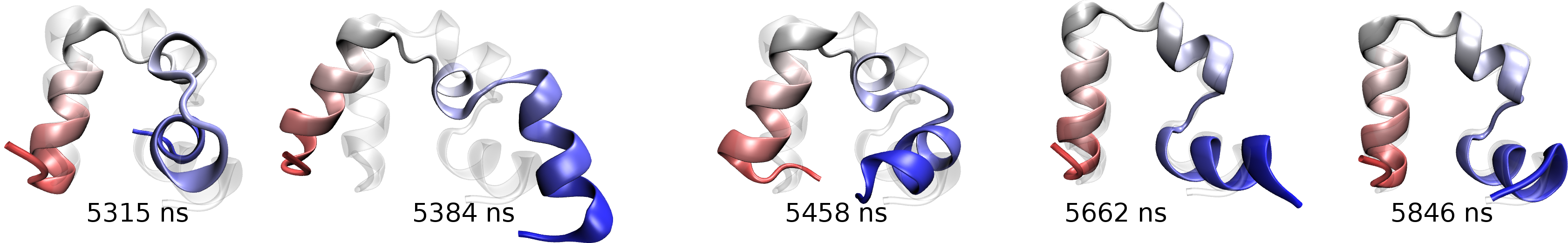 Key steps in the transition from the flipped to folded structure in a WT villin folding simulation. Click for a full size image.
Key steps in the transition from the flipped to folded structure in a WT villin folding simulation. Click for a full size image.
A fast-folding villin mutant
As noted above, fast-folding variants of villin headpiece have been isolated which are expected to fold in under one microsecond. In order to better characterize the folding of villin headpiece, we performed a series of six simulations on the fast folding mutant. Unexpectedly, the mutant showed more heterogeneous folding behavior than the wild type protein, in line with previous simulations by the Pande group using a different force field. Out of six trajectories, two folded (significantly faster than for the wild-type protein, in one to two microseconds), whereas the other four trajectories became trapped in misfolded states. While the failure of the fast-folding mutant simulations to reproduce the experimental folding times may indicate a problem with the force field (as in the case of the WW domain discussed above), it is also important to consider the observable that is experimentally used to measure folding. In the case of the villin headpiece, the experiments measure quenching of a tryptophan residue by a histidine residue one turn apart on helix III (the top of the red helix in the figure at left). In our simulations, however, we observed that even in the misfolding trajectories of the fast-folding villin mutant, the tryptophan-histidine distance reaches values equivalent to those in the native state after 1-2 microseconds, thus indicating that other observables may also need to be measured in order to reliably determine how quickly villin folds. Based on our simulations of both the wild type and fast-folding mutant villin headpiece, we were able to identify several salt bridges which are formed in all conformations except the native state, and a phenylalanine residue that only becomes involved in the hydrophobic core in the folded state. Both the formation of salt bridges and local environment of a phenylalanine are spectroscopically observable over the time scales of villin folding, and thus their measurement offers a concrete test for the vilin folding mechanism observed in our simulations. The presence of slower relaxation times in these observables than those found for, e.g., the currently used tryptophan quenching observable would both be evidence in favor of our model, and would provide a more accurate spectroscopic tool to measure the rate of villin headpiece folding.λ-Repressor
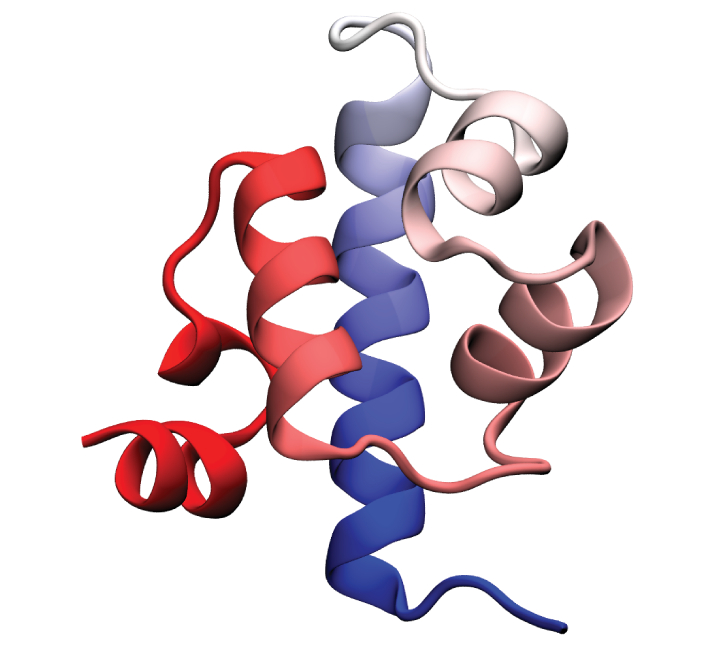 Folded conformation of the λ-repressor. Click to view a folding trajectory.
Folded conformation of the λ-repressor. Click to view a folding trajectory.
λ-repressor is another fast-folding protein we have studied using molecular dynamics simulations. The λ-repressor is more than twice the size of villin headpiece and processes a more complicated native topology, namely a five-helix bundle. Therefore, it is considerably more challenging and computationally intense to simulate than the previous cases (WW domain and villin headpiece). Despite its moderately large size, the experimentally known folding time for some of its mutants is less than 15 microseconds. Simulation of the λ-repressor in explicit solvent involves a system containing more than 74,000 atoms, and requires 10-20 microseconds to observe complete folding events.
Enhanced sampling simulation
We focused on a fast-folding mutant of the λ-repressor, λ-HG,
with a folding time of 15 microseconds in temperature-jump experiments.
By means of a recently developed tempering method (see Zhang and Ma, J. Chem. Phys. 132:244101 (2010)),
we observed reversible folding and unfolding of λ-repressor in a 10-microsecond trajectory (see movie at right). The folded state is
ranked as the most populated cluster without any prior knowledge
of the crystal structure in the subsequent cluster analysis.
Moreover, the pathway that leads to complete folding of the protein can be followed based on this cluster analysis.
Due to the enhanced sampling method used in the current study,
the folding pathways observed may not be the most probable ones.
Nevertheless, they represent one of the many physical pathways on the folding landscape.
In addition to accelerating the search in conformational space,
the enhanced sampling method also covers a broad range of temperatures in the simulation,
permitting the calculation of the temperature dependence of certain
structural characteristics given enough sampling (see figure below).
These results highlight the potential of this enhanced sampling method
and the accuracy of the underlying physical model (force field) in
studying a relatively large helical protein.
The simulations also revealed that the folding of λ-repressor is
not a simple two-state process as proposed for most fast-folding proteins.
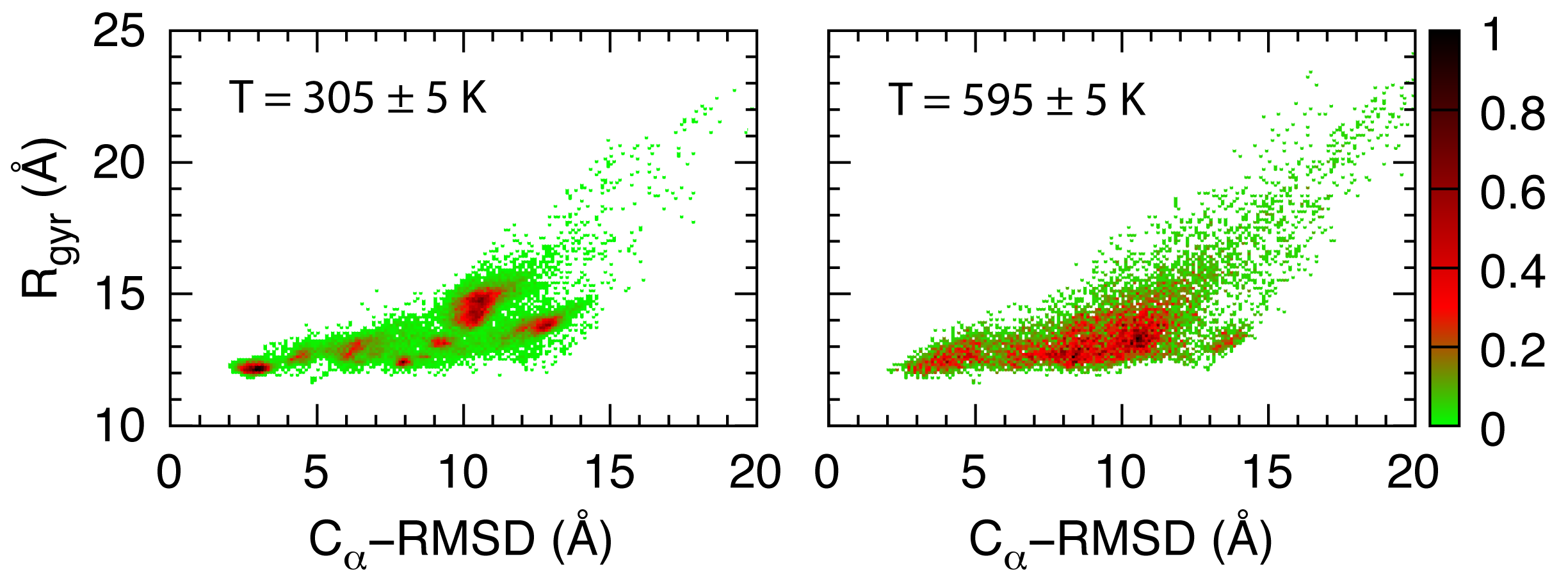 Temperature dependence of the histogram distribution of the radius of gyration and the root-mean-square-deviation.
Temperature dependence of the histogram distribution of the radius of gyration and the root-mean-square-deviation.
Constant temperature simulation
The enhanced sampling simulation permits the observation of the complete folding event but lacks information on the kinetics of the folding process. To obtain such information, constant-temperature simulations are required. We performed such simulations at three different temperatures: T = 329, 359, and 389 K (movies: 1, 2, 3). At T = 329 K, the protein accumulated more helical structure than that formed in the native state. The result may be due to the intrinsic bias towards helical structure in the force field. On the other hand, the helical overshot state may be analogous to the burst phase observed by circular dichroism kinetics measurements of λ-repressor in cryogenic solvent (see Dumont et al., Protein Sci. 15:2596 (2006)). At T = 389 K, the protein sampled several transient low-energy metastable state with native hydrophobic solvent accessible surface area, which confirmed that the folding of λ-repressor is a multiple-state process. The most interesting results came from the simulation at T = 359 K, with a folding trajectory as long as 0.1 millisecond with the help of a special purpose supercomputer, Anton. During this simulation, the protein partially folded with helices 1, 2, and 3 assuming their native conformation. Although helices 4 and 5 formed individually, their orientations relative to helix 1 are different from the ones in the native state. The observed folding pathway is different from the one being proposed before. The previous studies suggested that helices 1 and 4 orient correctly relative to the native structure more often than do helices 2 and 3. Moreover, the folding observed is slower than that measured in experiment. It is possible that the single trajectory in our simulation follows a variant, slow-folding pathway. We found a stable intermediate state, which has the small hydrophobic patch on helix 5 joining the main hydrophobic core formed by helices 1, 2, and 3. This stable intermediate slows down the folding in our simulation. The identification of the slow-folding pathway enables one to propose new experiment observables to better monitor the folding rate, as well as to propose possible fast-folding mutants that destabilized the observed intermediate state. Both predictions will be subject to future experiments to verify our simulation results.Pressure jump simulation
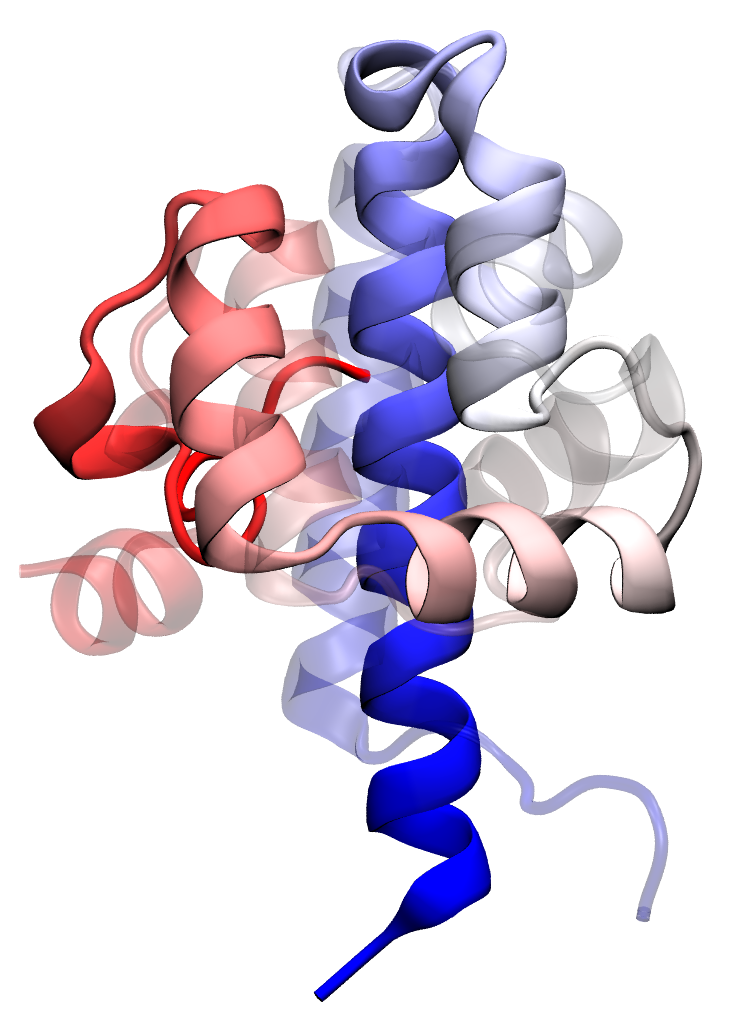 Transition state passage observed in a time window of 2 microseconds. Click to view a folding trajectory.
Transition state passage observed in a time window of 2 microseconds. Click to view a folding trajectory.
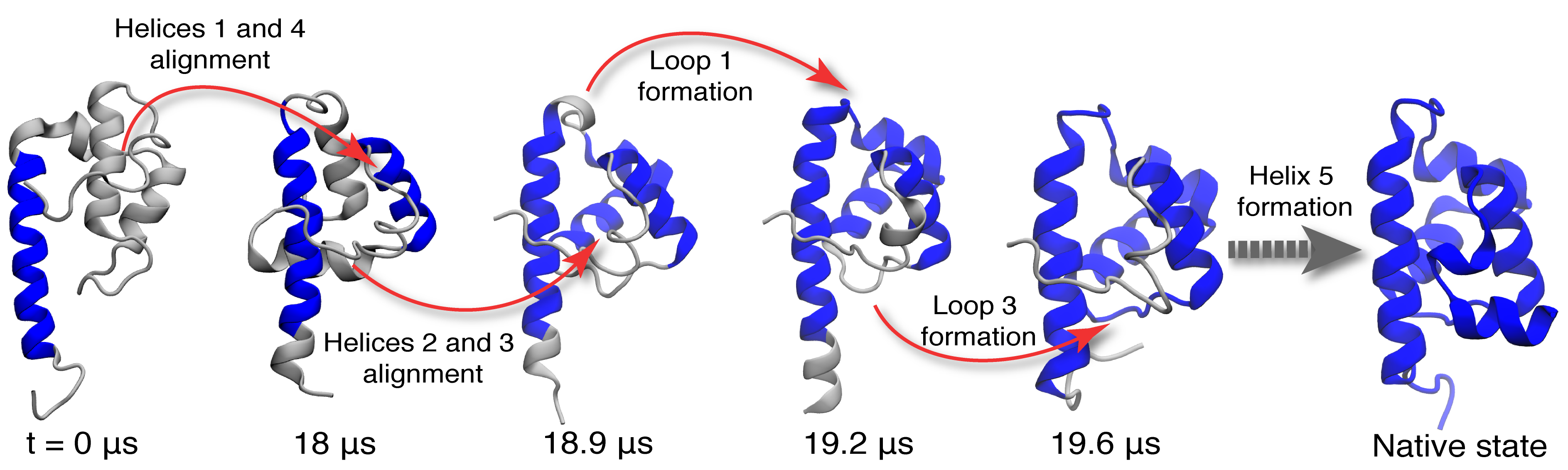 Molecular rearrangements observed in the simulation when the protein passes through transition states in the process of folding.
At each time point, the folded residues are colored blue.
Molecular rearrangements observed in the simulation when the protein passes through transition states in the process of folding.
At each time point, the folded residues are colored blue.
Publications
Investigators
Collaborators
Page created by Peter Freddolino and currently maintained by Yanxin Liu.
Last updated: May 28, 2015.


