Rafael C. Bernardi
Since August 2020, I am an Assistant Professor of Biophysics at the Department of Physics at Auburn University, and an external faculty member of the Theoretical and Computational Biophysics Group (TCBG). Before joining Auburn's faculty, I was a Postdoc (2012-2017), and then a Research Scientist (2017-2020), at the Beckman Institute at the University of Illinois. As part of the TCBG, I have been a member of the NIH Center for Macromolecular Modeling and Bioinformatics, which is known worldwide for the development of NAMD and VMD software, some of the most employed tools for computational structural biology, with approximately 300,000 users. Simultaneously, in 2017, I also held a Visiting Professor position at the chair of Applied Physics of the Ludwig-Maximilians-Universitat (LMU) Munchen, in Germany.
I have been very fortunate to work alongside some of the greatest minds of biophysics, including Prof. Klaus Schulten and Prof. Hermann Gaub, who inspired me to investigate some of the most complex biomolecular problems. Particularly, I am interested in protein mechanics, and how protein complexes behave under mechanical load. Such problem is very important in many areas of biomedicine, including bacterial adhesion during infection, and enzymatic activity in the gut microbiome. Also, I am interested in software development for computational structural biology, particularly in the use of hybrid QM/MM methods, and machine learning tools for analysis of molecular simulations. I am one of NAMD's developer, leading the efforts on the development of hybrid molecular mechanics/quantum mechanics (QM/MM) methods in both NAMD and VMD.
Home Department: Department of Physics at Auburn University.
Office Address: Leach Science Center, Room 2114.
Office Phone: (334) 844-4393
Email Address: rcbernardi@auburn.edu
Follow @rafaelcbernardiEducation
- PhD in Biophysics - Federal University of Rio de Janeiro - Brazil
- Masters in Physics - Brazilian Center for Research in Physics - Brazil
- BS Physics - State University of Londrina - Brazil
Advisor: Dr. Pedro Geraldo Pascutti and Dr. Werner Treptow
Advisor: Dr. Carlton Anthony Taft
Advisor: Dr. Andre Tsutomu Ota

5 Most Relevant Publications
1. LF Milles, K Schulten, HE Gaub***, RC Bernardi***; Molecular mechanism of extreme mechanostability in a pathogen surface anchor; Science, 359 (6383), 1527-1533, 2018
2. MCR Melo*, RC Bernardi*, T Rudack, M Scheurer, C Riplinger, JC Phillips, JDC Maia, GB Rocha, JV Ribeiro, JE Stone, F Neese, K Schulten, Z Luthey-Schulten***; NAMD goes quantum: A new integrative suite for hybrid simulations; Nature Methods, 15 (5), 351-354, 2018
3. RC Bernardi* ***, E Durner*, C Schoeler, KH Malinowska, BG Carvalho, EA Bayer, Z Luthey-Schulten, HE Gaub, MA Nash***; Mechanisms of Nanonewton Mechanostability in a Protein Complex Revealed by Molecular Dynamics Simulations and Single-Molecule Force Spectroscopy; Journal of the American Chemical Society, 141 (37), 14752-14763, 2019
4. C Schoeler*, RC Bernardi*, KH Malinowska, E Durner, W Ott; EA Bayer; K Schulten; MA Nash; HE Gaub***; Mapping mechanical force propagation through biomolecular complexes; Nano Letters, 15 (11), 7370-7376, 2015 - Cover Article
5. SM Sedlak*, LC Schendel*, HE Gaub***, RC Bernardi***; Streptavidin/biotin: tethering geometry defines unbinding mechanics; Science Advances, 6 (13), eaay5999, 2020
* equal contribution
*** corresponding author
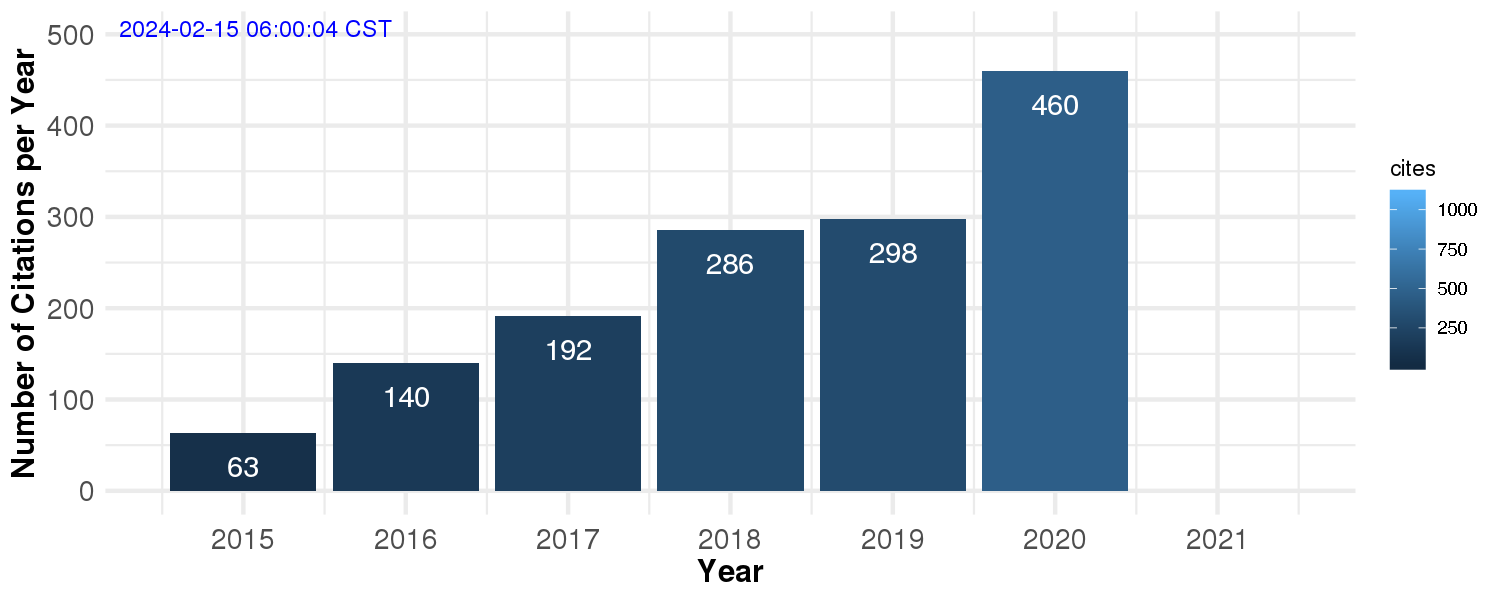
Number of Citations per Year (Google Scholar)
From 2014 to 2019 alone, I have published 17 original manuscripts with an average impact factor of approximately 12. Out of these 17 manuscripts, 9 of them were collaborations with experimentalists, with a highlight to a shared first author paper in Nature Methods in 2018, and my first senior author paper (last and corresponding author), which was published in Science, also in 2018. According to Google Scholar, my work has been cited over 1,000 times.




Research Interests
My research focus on the study of mechanoactive proteins using computational chemistry tools. Of note, mechanobiology is a vastly growing field, particularly due to the recent development of new experimental techniques, which have allowed us to better understand how cells sense and respond to mechanical stimuli, and, consequently, leverage this knowledge to develop new technologies for medical and industrial applications. Additionally, I am interested in software development for computational structural biology, particularly in the use of hybrid QM/MM methods, and machine learning tools for analysis of molecular simulations.
-
Mechanobiology
-
Protein Mechanostability
-
Force Propagation Analysis
-
Steered Molecular Dynamics
-
Single-Molecule Force Spectroscopy
-
Hybrid QM/MM Simulations
-
Machine Learning for MD analysis
-
Cellulose, Cellulases and Cellulosomes
-
Catch-Bonds
-
Gut Microbiome

Research Highlights
Research Highlights: Binding mechanism of Staph during human infection
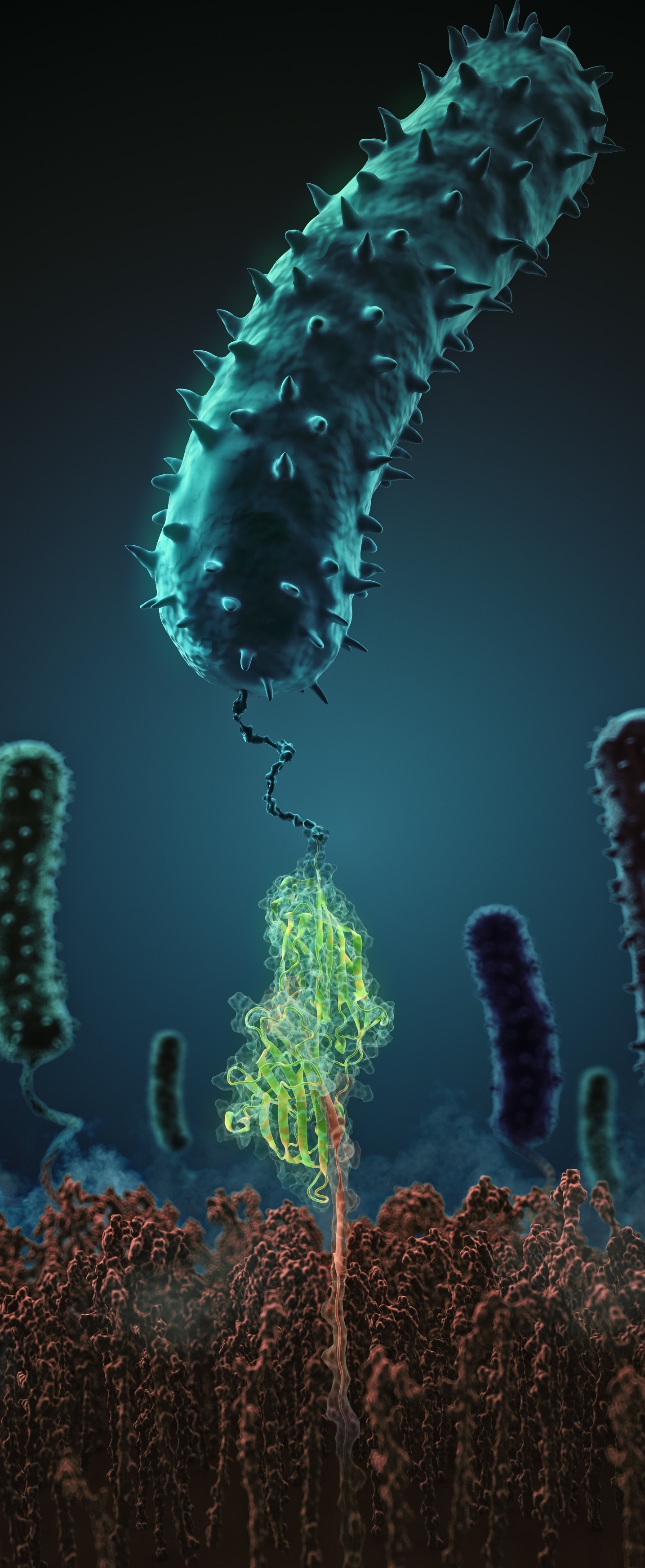
What makes pathogenic bacteria so persistent? Together with researchers from the University of Munich (LMU) we decipher the physical adhesion mechanism of a widespread pathogen virulence factor. Staph infections are caused by staphylococcus bacteria, types of germs commonly found on the skin of healthy individuals. Most of the time, these bacteria cause no problems or result in relatively minor skin infections. However, according to the Mayo Clinic, septicemia (also known as blood poisoning) can occur when staph bacteria enter a person's bloodstream. The bacteria can travel to locations deep within the body causing infections. Particularly vulnerable regions are medical devices such as artificial joints or cardiac pacemakers. Not surprisingly, staph infections are the leading cause of healthcare-related, so called nosocomial infections.
Staphylococcus epidermidis and Staphylococcus aureus are pathogens that can form biofilms on implants and medical devices. Central to biofilm formation is a very tight interaction between microbial surface proteins called adhesins and components of the extracellular matrix of the host. We used atomic force microscopy-based single-molecule force spectroscopy combined with steered molecular dynamics to explore how the bond between staphylococcal adhesin SdrG and its target fibrinogen peptide can withstand forces greater than 2 nanonewtons ( see the Perspective in Science by Herman-Bausier and Dufrêne ). The peptide is confined in a coiled geometry in a deep and rigid pocket through hydrogen bonds between SdrG and the peptide backbone. If pulled, the load is distributed over all hydrogen bonds so that all bonds must be broken at once to break the interaction. Read more in Science.
Our research in the news:
Research Highlights: QwikMD - Gateway to Easy Simulation

To assist experimentalists and any novice to MD to overcome the initial learning currve barrier of MD simulation software, we developed QwikMD, a user interface that connects the widely employed and user-friendly molecular graphics program VMD to the widely adopted MD program NAMD.
By enabling easy control of every step, QwikMD, meets even the needs of experts in the field, increasing the efficiency and quality of their work.
Get QwikMD now!
QwikMD offers:
Research Highlights: NAMD goes Quantum: Developing a new Modern QM/MM Interface
Hybrid methods that combine quantum mechanics (QM) and molecular mechanics (MM) can be applied to studies of reaction mechanisms in locations ranging from active sites of small enzymes to multiple sites in large bioenergetic complexes. By combining the widely used molecular dynamics and visualization programs NAMD and VMD with the quantum chemistry packages ORCA and MOPAC, we created an integrated, comprehensive, customizable, and easy-to-use suite (NAMD QM/MM). Through the QwikMD interface, setup, execution, visualization, and analysis are streamlined for all levels of expertise. Read more in Nature Methods.
Our research in the news:
Research Highlights: Second-generation Biofuels
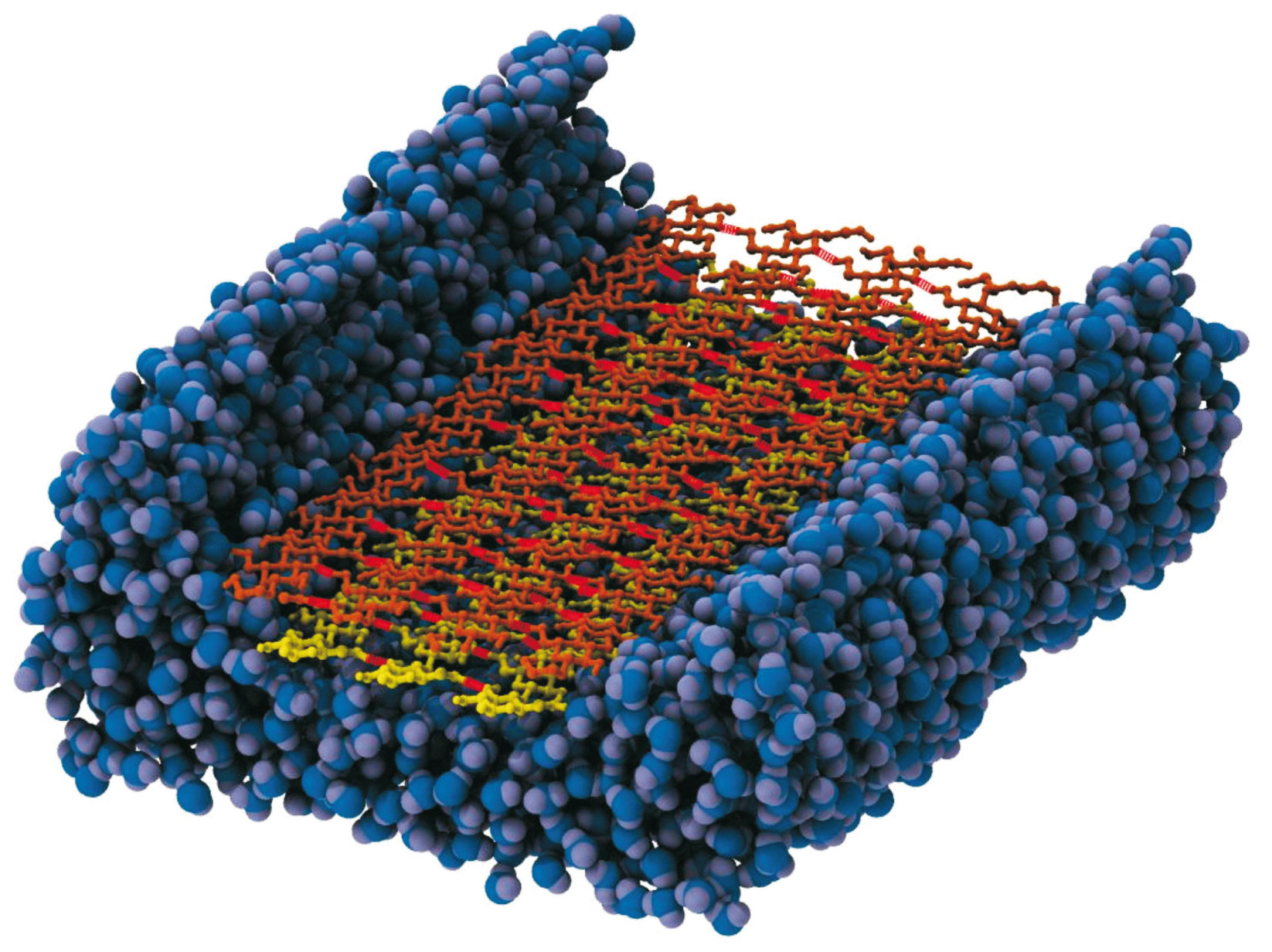
Biofuels are one of the most studied alternative sources of fossil-derived fuels, especially due to its smaller effect on greenhouse gas accumulation. The interest in the so-called second-generation biofuels has increased in the last few years, since they can be produced using the waste of agricultural production and thus do not raise ethical issues. Enzymatic hydrolysis to release carbohydrates units from plant cell wall polysaccharides has been one of the most promising strategies to produce the aforementioned new renewable biofuels. However the natural resistance of the plant biomass, usually referred to as biomass recalcitrance, makes the industrial process not cost-effective. To address this problem we are performing a joint computational and experimental study to unravel the mechanism of biomass degradation by cellulases, the enzymes found in some microbes that can digest plant fibers.
Cellulosomes
In another joint study we expect to elucidate the cooperative mechanism of cellulases complexes known as cellulosomes. The plant cell wall polysaccharides, cellulose and xylan represent the most abundant reservoirs of fixed carbon in nature and thus have considerable potential as a renewable energy source. These polysaccharides are highly recalcitrant to degradation; however fungi and bacteria have evolved complex enzymatic strategies for the degradation and fermentation of these polysaccharides. One of the important paradigms for enzymatic degradation of plant cell walls is the cellulosomal system, elaborated by certain strains of bacteria. These extracellular proteinaceous assemblages consist of a scaffoldin backbone onto which cohesin-containing catalytic modules and carbohydrate binding modules are appended. Analogous to a Swiss army knife, these cellulosomes contain a plethora of different catalytic and substrate binding activities that facilitate the degradation of plant cell wall material. It was reported that Clostridium thermocellum, the most studied cellulosomal organism, exhibits one of the highest rates of cellulose utilization known in nature, and the cellulosomal system of this bacterium is reported to display a specific activity against crystalline cellulose that is 50-fold higher than the corresponding non-cellusomal fungal system in Trichoderma reesei. To understand the efficient mechanism that leads cellulosomal machineries in organisms to degrade biomass, we are studying the interaction of the subunits of the cellulosome to comprehend their synergistic mechanism.
Research Highlights: One of the Strongest Biomolecular Interactions

Bacteria can make a living from a very wide range of food sources. This ability makes them, for example, essential symbionts in animal digestive tracts where they assist their hosts in breaking cellulose fibers up into compounds degradable by the animal metabolism. Today, human gut bacteria, part of the human microbiome, are one of the hottest research topics in medicine. Gut bacteria face a particularly tough job in the rumen of the cow where they digest hardy cellulose fibers of grasses. Key to the job, taking place in a constantly moving fluid, are molecular tentacles, so-called cellulosomes, on the surface of the symbiotic bacteria. The cellulosomes develop a tight grasp on and then effective cleavage of cellulose. In a joint experimental-computational study researchers have investigated how in case of the bacterium Ruminococcus flavefaciens cellulosomes are built in a modular way, with molecular modules easily binding and unbinding during cellulosome construction, but sticking extremely strongly together during cellulosome digestive activity. As reported recently, single molecule force microscopy and molecular dynamics simulations using NAMD could show that under strain the adhesive bonds between cellulosome modules become stronger than seen in any other biomolecular system, in fact, become nearly as tight as strong chemical bonds. While the experimental data revealed bond strength and other characteristics, simulations reproducing the observed data provided a detailed view of the adhesive bond at atomic resolution, thereby revealing the physical mechanism underlying the uniquely adhesive property of cellulosomes. Gut bacteria and cellulosomes can be employed in 2nd generation biofuel production. Read more from the news below or from our Nature Communications and Nano Letters manuscripts.
Our research in the news:
Previous Projects
Studying membrane lipids in the DFT level using a QM/MM approach
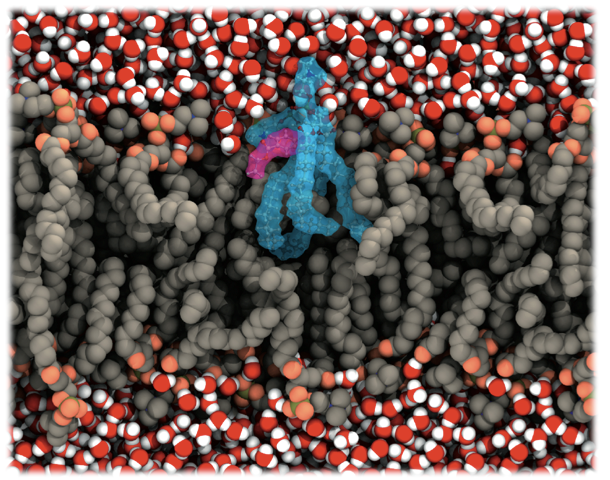
From very simple models, such as dielectric interfaces in the 1980s and 1990s, to all-atoms simulations with thousands of lipids, going through several methods and models developed, biological membranes as well as their proteins have become one of the most studied systems by molecular dynamics simulations, especially because of the huge advances in the computer capability. Membranes are especially important to drug development, since several membrane proteins are drug targets. Moreover, the membrane is a remarkable barrier to a majority of the drugs that act inside the cell.
Numerous studies attempt to understand how could we better use computational methods to study such regions, for instance, using more accurate methods of molecular modeling, such as quantum mechanical (QM) calculations. Meanwhile, because this interface has electrostatic properties that, at first, could not be studied by classical molecular modeling, the application of QM methods seems to be a more accurate approach. Furthermore, the behavior of the charge distribution over a drug surface, especially those containing aromatic groups, will adapt to the media when crossing a membrane interface, opposite of the classical MD approach, where it remains constant.
To study biological systems, especially enzymatic reactions, a hybrid QM/MM approach in molecular dynamics have been largely used. However, despite its importance in several biochemical processes, to the best of our knowledge, the very same approach has not been used to study the lipid membranes interface using a quantum mechanical treatment for the lipids. Understand how amphiphilic drugs interact, at the level of molecular orbitals, with the lipids in this region is essential for the complete knowledge of such drugs mechanism.
We choose to study how the local anesthetic benzocaine behaves in such region, once its mechanism of action may be related to its stabilization on the membrane/water interface. The QM/MM simulations were carried out taking advantage of an integration of the Car-Parrinello method, to treat the benzocaine as well as two of the membrane lipids and the GROMOS force field to simulate the remaining system. Our calculations showed an overlap of the atomic orbitals of the anesthetic and the lipid, suggesting a strong hydrogen bond between the amine terminal of the drug and the palmitate group of the lipid.
Protein Folding
One of the main paradigms of molecular biology tells us that the three-dimensional structure of proteins defines its function and dynamics. Such three-dimensional structures, in turn, derive from the amino acid sequence itself, through the folding process. A protein's structure determines its activity and properties, thus knowing such conformation on an atomic level is essential for both basic and applied studies of protein function and dynamics. However, the acquisition of such structures by experimental methods is slow and expensive, and current computational methods mostly depend on previously known structures to determine new ones. We developed a new software called GSAFold that applies the Generalized Simulated Annealing (GSA) algorithm on ab-initio protein structure prediction. The GSA is a stochastic search algorithm employed in energy minimization and used in global optimization problems, especially those that depend on long-range interactions, such as gravity models and conformation optimization of small molecules. This new implementation applies, for the first time in ab-initio protein structure prediction, an analytical inverse for the Visitation function of GSA. It also employs the broadly used NAMD Molecular Dynamics package to carry out energy calculations, allowing the user to select different force fields and parameterizations. Moreover, the software also allows the execution of several simulations simultaneously. Applications that depend on protein structures include rational drug design and structure-based protein function prediction.


