Force Field Toolkit Plugin, Version 2.1
Overview
The Force Field Toolkit is comprised of a set of tools that aid users in the development
of CHARMM-compatible forcefield parameters, including charges, bonds, angles, and dihedrals.
These tools are accessed through the provided GUI, which greatly simplifies the setup and
analysis of the underlying calculations.
Amber-compatible parameters can also be generated, although Amber-style force field files are not
output (users can convert though with parmed).
Currently, the toolkit assumes that all QM target
data is generated using Gaussian; however, we plan to expand functionality to include alternatives.
For a full description of ffTK, please see:
C.G. Mayne, J. Saam, K. Schulten, E. Tajkhorshid, J.C. Gumbart.
J. Comput. Chem.
2013, 34, 2757-2770.
DOI: 10.1002/jcc.23422.
To get the most out of the ffTK, a reasonable understanding of the CHARMM and CGenFF force fields is required. For additional information on these topics and the underlying principles of parameterization, users are urged to also visit Alex MacKerell's website for CHARMM FF Parameters and the CHARMM user forums, specifically the parameterization section.
The ffTK plugin relies on a number of other VMD plugins, described in more detail here: Multiplot, NAMD Energy, Optimization, QM Tool, Topo Tools
** Update Notice **
ffTK has been significantly updated for version 2 to include bug fixes and support for ORCA.
We strongly recommend using the latest available version of ffTK, which is distributed as part of
VMD 1.9.4.
For the most cutting edge features download the latest VMD alpha release.
Authors
Christopher G. Mayne, Programmer, Beckman Institute for Advanced Sciences and Technology, University of Illinois at Urbana-ChampaignJames C. Gumbart, Assistant Professor of Physics, Georgia Institute of Technology
Other Contributors
Hongde Yu (undergraduate student, Tsinghua University, Beijing, China) contributed to the development of tools for parsing, processing, and analyzing CGenFF/ParamChem output.Mark Wetherington (MS 2014, Georgia Tech, Atlanta, GA) contributed to the development of tools for charge calculations using RESP.
How To Cite ffTK
When publishing work that has used ffTK, please cite the following papers:

Rapid parameterization of small molecules using the Force Field Toolkit
J. Comput. Chem., 2013, 34, 2757-2770.
Cover Article, Top-Ten Most-Accessed Article 2014
Tutorials
We have developed training materials to guide users through the entire parametrization process for a small organic molecule, pyrrolidine. The full workflow is broken into modular steps, each with a specififc focus on the technical aspects of using ffTK. Additional information pertaining to the theory behind pyrrolidine parametrization can be found elsewhere (Vanommeslaeghe, K. et al. J. Comput. Chem. 2010, 31, 671-690).
A complete set of tutorial materials (i.e., PDF, sample files) can be found on the Tutorials website under "Specialized Topics": Parameterizing Small Molecules Using the Force Field Toolkit (ffTK).
A screencast version of the tutorial can be found on the ffTK Research page.
GUI Documentation
BuildPar
BuildPar is a collection of three tools for identifying missing parameters, building an initial parameter file, and updating the new parameter file based on ffTK output.
- ► Identify Missing Parameters
- Description:
- The Identify Missing Parameters tool determines all of the parameters necessary to describe the given molecule, cross-checks the required parameters against established parameter files, and constructs a new CHARMM-compatible parameter file containing only the missing parameters, for which all values are set to zero.
- Usage:
- Set the inputs appropriately. Run the tool using the Write Initial Parameter File button.
- Input:
- Input PSF File: A PSF file for the molecule of interest; atoms must be appropriately typed.
- Associated Parameter Files: CHARMM-formatted parameter file(s) that will be searched against.
- Output PAR File: A filename for the output zeroed parameter file.
- Output:
- A CHARMM-compatible parameter file containing any missing parameters (bonds, angles, dihedrals, non-bonded). All values are set to zero.
- ► Assign Missing VDW/LJ Parameters by Analogy
- Description:
- ffTK does not currently support the optimization of non-bonded parameters; however,
new atom types will require such parameters. The Assign Missing VDW/LJ Parameters
by Analogy tool is designed to aid the user in assigning missing non-bonded
parameters by analogy from established parameter sets. The tool is divided into two
panes: the top pane is for loading the initial parameter file that the user is developing,
and the bottom pane is for browsing non-bonded parameters read in from established parameter
files.
Important Note: The parameter browser requires that the user load BOTH the topology AND parameter files for a given parameter set. This is because the topology file often contains information in the comments that helps identify properties of a given atom type. - Usage:
- Enter the location/file name of the initial parameter file. Read in any missing non-bonded
parameters using the Load button. After the parameters are properly set, the values are
updated in the parameter file using the Update File button.
Reference parameter sets are loaded from the Load Topology + Parameter Set button, which launches a dialog requesting the location and filename of a topology and parameter file pair. Any non-bonded parameters found within the file pair are loaded into the browser box, providing the element, atom type, parameter values, and the file name from which they came. Multiple parameter sets can be loaded sequentially. Parameters can be restricted to certain elements or parent files using the drop down menus.
To assign the parameters in the input parameter file, select both the target entry from the top pane, and desired reference entry from the bottom pane. The Set from Reference button will copy the reference parameter values to the target entry. - Input:
-
- Incomplete PAR File: The parameter file containing the missing VDW/LJ parameters (e.g., the output PAR file from Identify Missing Parameters tool).
- Topology + Parameter Set: The matching topology and parameter files from which VDW/LJ parameters will be chosen.
- Output:
- The Update File button overwrites the Incomplete PAR File. No new files are generated.
- ► Prepare Parameterization from CGenFF Program Output
- Description:
- Entry into the ffTK workflow minimally requires a PSF/PDB file pair, which can be used to generate an initialized (zero-filled) parameter file, as described above (see Identify Missing Parameters). An externally developed resource named the CGenFF Program can provide a CHARMM stream file containing topology information and initial parameters assigned by analogy to small molecules present in the standard CGenFF (the force field) distribution. The tool described here aides users in processing output from the CGenFF Program to write a properly formatted PSF/PDB file pair, write an initial parameter file based on analogy rather than zero-filling, and provides ability to inspect penalty terms associated with each parameter and where each term is found in the molecule.
- Usage:
- Set the inputs appropriately. The first step is to analyze the input data, which will load the molecule into VMD and populate the parameter data. After analysis, the data can be used to write a PSF/PDB file pair and/or an initial parameter (PAR) file that serve as input to the ffTK workflow. Selecting a parameter entry, or multiple entries, will draw graphic objects that identify each occurrence of the selected parameter within the molecule in the VMD OpenGL window.
- Input:
-
- Input PDB/MOL2: The structure file submitted to the CGenFF Program.
- CGenFF STR File: The CHARMM stream file returned by the CGenFF Program.
- Output Folder: The folder where output data will be written.
- Resname: Residue name for the molecule. All atoms in the input PDB/MOL2 will be assigned to this resname. The resname will also serve as the basename for all output (i.e., PSF/PDB, PAR).
- Chain: Chain identifier used when writing PSF/PDB output. Defaults to L.
- Segment: Segment identifier used when writing PSF/PDB output. Defaults to L.
- Output:
- The Write PSF/PDB button will write the structure to PSF and PDB files. The Write PAR button will write out a parameter file. If the CGenFF stream file contains all parameters required to describe the molecule (an option available when submitting input to the CGenFF Program), the parameters are split into two files denoted as "existing" and "missing". In subsequent ffTK steps, the "existing" file would be characterized as an "associated parameter file." All output is written to the specified output folder and will use the provided resname as the basename for each file.
- ► Update Parameter File with Optimized Parameters
- Description:
- The Update Parameter File with Optimized Parameters tool serves as the primary mechanism for assigning optimized parameter values in the developing parameter file from ffTK output files (LOG).
- Usage:
- The user provides an input parameter file for updating, the ffTK LOG file containing the relevant output, and a file name for the new (updated) parameter file. The tool is run via the Write Updated Parameter File button.
- Input:
- Input Parameter File: The parameter file currently under development.
- Optimization LOG File: ffTK output file (LOG) from the calculation or optimization.
- Output Parameter File: A location and file name for the resulting output parameter file.
- Output:
- A parameter file containing updated values based on the input.
Opt. Geometry
- Description:
- The Opt. Geometry tab is designed to simplify the process of obtaining a QM-optimized structure for a given molecule.
- Usage:
- The top section of the tool serves to write the Gaussian input file (GAU), while the bottom section allows the user to visualize the optimization steps and write out a final structure to a PDB file.
- Input:
- Input PDB File: The location and filename of PDB file containing the initial coordinates.
- Output GAU File: A location and filename for the generated Gaussian input file.
- Gaussian Settings: The default Gaussian settings are for a geometry optimization at the MP2/6-31G* level of theory for a neutral molecule, and should be suitable for most cases.
- Original PDB File: The location and filename of the PDB file used as the Input PDB File above.
- Gaussian LOG File: The location and filename of the LOG file generated from the Gaussian calculation.
- Output PDB File: A location and filename for the output PDB file containing the QM-optimized atomic coordinates (geometry).
- Output:
- A Gaussian input file (GAU) for running the Gaussian calculation.
- A PDB file containing the QM-optimized atomic coordinates (geometry).
CHARMM: Water Int.
- Description:
- The Water Int. tab is designed to aid the user in generating the QM water interaction target data used in charge optimization.
- Usage:
- The user provides the requested input information regarding the molecule, followed by loading the molecule into VMD (Load PSF/PDB button). The Hydrogen Bonding Atoms box provides several tools for assigning donor and acceptor indices. These tools include, atom labeling, colored spheres for each listed index, and a routine that autofills the donor/acceptor lists based on a relatively simple algorithm. Default settings for the Gaussian calculation are provided in the Gaussian Settings box. The Write Gaussian Input Files button generates a Gaussian input file (GAU) that optimizes the interaction of a water molecule with a specific atom, provided in the donor/acceptor lists. The resulting GAU file(s) should be checked visually by loading them into VMD via the Load GAU Files button. After the Gaussian calculations have completed, the user can visualize the optimization steps by loading the LOG file(s) generated from each Gaussian calculation via the Load LOG Files button.
- Input:
- PSF File: The location and filename of the PSF file describing the molecule.
- PDB File: The location and filename of the PDB file containing the QM-optimized geometry.
- Output Path: The location where GAU files will be written.
- Basename: A short abbreviation used for naming GAU files; generally the residue name.
- Donor Indices: The index for each atom that will positively interact with the oxygen atom of a water molecule.
- Acceptor Indices: The index for each atom that will positively interact with the hydrogen atom of a water molecule.
- Gaussian Settings: The default Gaussian settings optimize the distance and orientation of the water molecule with respect to a given atom in the molecule at the RHF/6-31G* level of theory. These settings should be suitable for most cases.
- Output:
- A Gaussian input file (GAU) for each index provided in the donors/acceptors lists.
CHARMM: Opt. Charges
- Description:
- The Opt. Charges tab is used to setup and run a calculation that optimizes atomic partial charges to reproduce water interaction QM target data using MM calculations. The optimization requires a number of settings, which are broken down into sections that will be described separately.
- Usage:
- Appropriate data are assigned for Input, Charge Constraints, and QM Target Data sections. The optimization
is launched by the Run Optimization button, whereby the status label will update periodically
during the calculation. Upon completion, the status returns to
IDLE
and the optimized charge data is loaded into the Results section. A new PSF file containing the optimized charges can be written by specifying a location and filename in theUpdate PSF with new charges
box, and clicking the Write button. Note that writing a new PSF requires that the PSF and PDB entries are set in the Input section. The results from previous charge optimizations can be reloaded by specifying the ffTK output log file intoLoad output file from a previous optimization
and selecting the Load button. - Input/Settings:
- ► Input
- PSF File: The location and filename of the PSF file describing the molecule.
- PDB File: The location and filename of the PDB file containing the QM-optimized geometry.
- Residue Name: The VMD
resname
abbreviation for the molecule. - Parameter Files: Any parameter files that are required for simulation of the molecule. This includes associated parameter files, as well as the initial parameter file that is under development.
- NAMD binary: The location of NAMD binary file, or simply the filename if the location is included in the PATH.
- Output LOG: A location and filename for ffTK output log file (LOG).
- ► Charge Constraints
- Charge Group: A list of atom names that will be forced to have the same charge.
- Initial Charge: An initial charge assigned to the charge group. Cannot be zero.
- Low Bound: The lowest charge that can be assigned to the charge group.
- High Bound: The highest charge that can be assigned to the charge group.
- Net Charge: Integer charge for the entire molecule, e.g., 0 for a neutral molecule.
- ► QM Target Data
- Cmpd LOG (HF): The location and filename of the Gaussian log file containing the single point energy calculated for the molecule at the same level as the water interaction (e.g., RHF/6-31G*).
- Cmpd LOG (MP2): The location and filename of the Gaussian log file containing the single point energy calculated for the molecule at a high level of theory (e.g., MP2/6-31G*) to obtain an accurate dipole moment.
- Water LOG: The location and filename of the Gaussian log file containing the single point energy calculated for a TIP3 water molecule calculated at the same level as the water interaction (e.g., RHF/6-31G*).
- LOG File: Log file for each QM-calculated water interaction.
- Atom Name: The atom name for the atom specified in the water interaction calculation.
- Weight: A weighting factor for the associated water interaction data.
- ► Advanced Settings
- Start, End, Delta: Control how the water molecule is moved with respect to the specified atom during the MM calculation. Values are relative to the QM-optimized water interaction.
- Offset: Adjustment applied to distance of the water molecule at the QM energy minimum to account for differences between gas-phase and bulk-phase calculations .
- Scale: Scaling factor applied to the QM minimum energy to account for the difference between gas-phase and bulk-phase calculations.
- Tolerance: Tolerance used to determine the termination criertia for the optimization function.
- Dist. Weight: Weighting factor of QM Distance relative to QM energy. Default is 1.0. Values < 1.0 weight energy more heavily and are reasonable, if not desired.
- Mode: Defines how the optimizer runs; downhill and simulated annealing are supported (see Optimization for details).
- Override ReChargeFromPSF: If checked, replaces charges from PSF with those provided for the specified atom name during the optimization. Note, that
Optimized Sum
will need to be recalculated via the Calculate from PSF button after modifying this setting. The syntax is: {atom_name charge} for each atom specified. - Number of Iterations: This option allows one to preform multiple rounds of charge optimization automatically, using the output of each round as the input of the next round.
- Write debugging log: If checked, output also includes a .debug.log file that contains detailed information about the setup and calculation.
- Build Run Script: If checked, instead of running the optimization immediately, ffTK generates a Tcl script with all of the commands necessary to setup and run the optimization. This is especially useful for running the optimization with VMD in text mode. Note that input files are assigned with absolute paths, and may need to be adjusted if the script is launched from a machine with a different filesystem.
- ► Results
Charge groups should only contain atoms for which charges will be optimized. Each group can be added manually, orguessed
using an algorithm that assigns charge groups based on chemical equivalency if the PSF/PDB has been loaded into VMD as the TOP molecule (see Input section). The Input section also has a tool to show a variety of labels for each atom to aid in assigning charge groups. The Calculate from PSF button calculates theOptimized Sum
using atomic charges excluded from the optimization found in the PSF, and/or charges that are manually specified via the "Override ReChargeFromPSF" box under Advanced Settings.
The geometry optimization is typically calculated at MP2/6-31G*, and therefore, a separate single point energy calculation of the optimized geometry at RHF/6-31G* is required for the Cmpd LOG. The Water Interaction Energy Data are loaded via the add button. If the water interaction energy data were generated using the Water Int. tab, then the Atom Name will be automatically parsed from the filename and set accordingly. One exception to this is for water interaction files probing a carbonyl, which will need to be modified to give the appropriate atom name due to a difference in the default GAU file naming scheme.- The results provide the optimized charge for each charge group.
The
Charge Total
should match theOptimized Sum
as calculated in Charge Constraints section; however, rounding errors can occur. In such cases, select a charge group entry and adjust the charge as necessary, taking the number of atoms in the charge group into account. Updating the PSF and loading other outputs are discussed inUsage
above. It is strongly suggested that users employ the Charge Optimization Log Plotter (COLP) utility to assess optimization convergence and the performance of the resulting charges in the ability to reproduce the QM target data.- Output:
- A ffTK log file for use in updating charges via the Update PSF with new charges in the Results section.
Amber: Calc. ESP
- Description:
- The Calc ESP tab is designed to aid the user in setting up a Gaussian calculation of the Electrostatic Potential.
- Usage:
- The user provides the Gaussian checkpoint file from a previous optimization. Default settings for the Gaussian calculation are provided in the Gaussian Settings box. The Write Gaussian Input File button generates a Gaussian input file (GAU) for calculation of the molecule's electrostatic potential.
- Input:
- Geometry Optimization CHK File: The location and filename of the Gaussian checkpoint file generated during the geometry optimization. Note: Calc. ESP makes a copy of this file, preventing an overwrite of the original by Gaussian.
- Output GAU File: A location and filename for the generated Gaussian input file.
- Gaussian Settings: Default settings are provided for calculation of the electrostatic potential at HF/6-31G* level of theory.
- Output:
- A Gaussian input file (GAU) for calculating the electrostatic potential.
- A Gaussian checkpoint file using during the ESP calculation.
Amber: Opt. ESP
- Description:
- The Opt. ESP tab is used to setup and run a calculation that optimizes atomic partial charges using the "resp" program distributed as part of AmberTools to fit the electrostatic potential. The optimization requires a number of settings, which are broken down into sections that will be described separately.
- Usage:
- Appropriate data are assigned for Input and Charge Constraints sections. The calculation
is launched by the Calc. RESP Charges button. Upon completion, the status returns to
IDLE
and the new charges are written to an updated PSF file. - Input/Settings:
- ► Input
- PSF File: The location and filename of the PSF file describing the molecule.
- PDB File: The location and filename of the PDB file containing the QM-optimized geometry.
- Residue Name: The VMD
resname
abbreviation for the molecule. - Net Charge: Integer charge for the entire molecule, e.g., 0 for a neutral molecule.
- ► Charge Constraints
- Charge Group: A list of atom names that will be forced to have the same charge.
- Initial Charge: An initial charge assigned to the charge group.
- Restraint Type: If set to Static, the charge will not be changed. If set to Dynamic, all charges in the list will be restrained to the same final value (Note: the list must contain more than one atom). If set to none, the atom will be treated normally.
- Restraint Atom: The atom of the group to which others will be restrained. This should default to the first atom of the list.
- ► Input File Settings
- Gaussian ESP Log: The location and filename of the Gaussian log file containing the electrostatic-potential calculation.
- Input Files Basename: A short abbreviation used for naming resp input files; generally the residue name.
- ihfree: If ihfree = 0 all atoms are restrained. If ihfree = 1, hydrogens are not restrainted (default behavior).
- qwt:Weighting factor (default is 0.0005).
- iqopt: Determines initial charges. If iqopt = 1, all initial charges are set to 0; if iqopt = 2, initial charges are read in (default).
- ► Calculate RESP Charges
- Updated PSF Name: The name of the PSF that will contain the updated charges determined by resp.
- RESP path: The location of the resp program, or simply the program name if the location is included in the PATH.
All atoms should be listed in this window; if a given charge will not be optimized, it should be set to Static. Each group can be added manually, orguessed
using an algorithm that assigns charge groups based on chemical equivalency if the PSF/PDB has been loaded into VMD as the TOP molecule (see Input section). Charges can be added to groups or split from groups.
Calc. Bonded
- Description:
- The Calc. Bonded tab is used to setup the Gaussian calculation of the hessian used to optimize bonds and angles.
- Usage:
- The user provides the location and filename of the Gaussian checkpoint (CHK) file generated during the geometry optimization (Opt. Geometry), and a filename for a Gaussian input file to calculate the hessian, which is written using the Write Gaussian Input File button.
- Input:
- Gaussian Settings: Default settings are provided for calculation of the hessian at MP2/6-31G* level of theory.
- Geometry Optimization CHK File: The location and filename of the Gaussian checkpoint file generated during the geometry optimization. Note: Calc. Bonded makes a copy of this file, preventing an overwrite of the original by Gaussian.
- Output GAU File: A location and filename for the generated Gaussian input file.
- Output:
- A Gaussian input file for running the hessian calculation.
- A Gaussian checkpoint file used during the hessian calculation.
Opt. Bonded
- Description:
- The Opt. Bonded tab is used to setup and run a series of calculations that optimize bonded force constants and equilibrium values against QM target data using MM calculations. The optimization requires a number of settings, which are broken down into sections that will be described separately.
- Usage:
- Appropriate data are assigned for Input and Parameters to Optimize sections. The optimization is launched by the Run Optimization button,
whereby the status label will update periodically during the calculation. Upon completion, the status returns to
IDLE
and the optimized bonded parameters are loaded into the Results section, along with a final objective value reached by the optimizer. The optimization can be run iteratively, and upon satisfactory results, the in-progress parameter file is updated using the BuildPar tab described above. - ► Input:
- PSF File: The location and filename for the PSF file describing the molecule.
- PDB File: The location and filename for the PDB file describing the optimized geometry of the molecule.
- Hess LOG File: The location and filename for the LOG file generated from the Gaussian calculations.
- In-Progress PAR File: The location and filename for the parameter file containing unparameterize bonds and angles.
- Additional Associated Parameter Files: The location and filenames for any PAR files, other than the in-progress file, that contains parameters required to describe the molecule.
- NAMD Bin: The location of the NAMD executable, or simply its name if the location is included in the PATH
- Output LOG: The location and filename for writing an ffTK output log file (LOG).
- ► Parameters to Optimize:
- Bond/Angle: Type of parameter, bond or angle.
- Atom Type Def.: The parameter definition using atom types; space delimited.
- Force Constant: An initial guess for the force constant.
- b0/θ: An initial guess for the equilibrium value (Å or degrees).
- ► Advanced Settings:
- Tolerance: Tolerance used to determine the termination criteria for the optimization function.
- Geom. Weight, Energy Weight: Weighting factors for tuning the relative contribution of geometry and energy terms to the objective function. Defaults are 1.0.
- Mode: Defines how the optimizer runs; downhill and simulated annealing are supported (see Optimization for details).
- Eq. Deviation: Threshold for nonpenalized deviation from the target equilibrium value.
- K Lower Bound, K Upper Bound: Bounds applied to each force constant. Bonds and angles have separate bounds.
- Write debugging log: If checked, output also includes a .debug.log file that contains detailed information about the setup and calculation.
- Build run script: If checked, instead of running the optimization immediately, ffTK generates a Tcl script with all of the commands necessary to setup and run the optimization. This is specifically useful for running the optimization with VMD in text mode. Note that input files are assigned with absolute paths and may need to be adjusted if the script is launched from a machine wiht a different filesystem.
- ► Results:
- The results provide the optimized force constant and equilibrium value for each bond/angle parameter entry, along with the final objective value reached by the optimizer.
- Output:
- An ffTK log file for use in updating bond and angle parameters in the parameter file under development using BuildPar.
Scan Torsions
- Description:
- The Scan Torsions tab is designed to aid the user in generating the QM target data used to optimize dihedral parameters from torsion scans.
- Usage:
- The user provides required input/output information. Loading the PSF/PDB allows the user access to several tools for selecting dihedral angles to be scanned. The preferred method for adding entries to the Dihedrals to Scan box is via the Read from PAR button. This button uses a dialog to identify the parameter file under development, from which dihedrals missing parameters are read, and non-redundant torsions are added to the box. Each dihedral can be visualized in VMD by selecting the entry. Dihedrals can also be added manually via the add button. The Generate Dihedral Scan Input Files button generates two Gaussian input files per scan, one in the positive direction and one in the negative direction.
- Input:
- PSF File: The location and filename for the PSF file describing the molecule.
- PDB File: The location and filename for the PDB file describing the QM-optimized molecular geometry.
- Output Path: The location where Gaussian input files are written.
- Basename: A short abbreviation used for naming GAU files; generally the residue name.
- Dihedral Atoms: Indices describing torsion to scan.
- Scan +/-: Range to scan from Equilibrium Value in either direction (degrees).
- Step Size: Distance between scan steps (degrees).
- Gaussian Settings: The default Gaussian settings perform a relaxed potential energy scan at the MP2/6-31G* level of theory. These settings should be suitable for most cases.
- Output:
- Two Gaussian input files for each entry of the Dihedrals to Scan box.
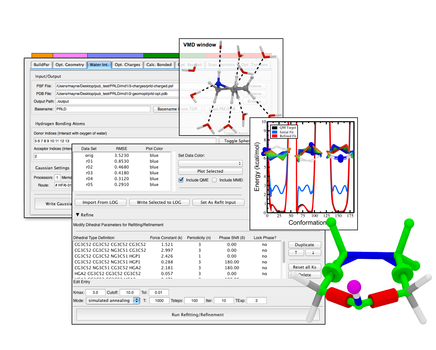
Opt. Torsions
- Description:
- The Opt. Torsions tab provides the tools to setup and run calculations that optimize dihedral angle parameters to reproduce QM relaxed potential energy scans. The optimization requires a number of settings, which are broken down into sections that will be described separately.
- Usage:
- Appropriate data are assigned for Input, QM Target Data, and Dihedral Parameter Settings sections. The initial optimization
is launched by the Run Optimization button, whereby the status label will update periodically during the calculation.
Upon completion, the status returns to
IDLE
and optimization data is loaded into the Visualize Results section. Here, the data can be plotted against the QM target data (QME) and the initial MM data that excludes energy contributions from the dihedrals being fit (MMEi). The initial optimization usually requires refinement through the use of the Refine section. Initial optimization is copied to the Refine section by selecting the entry in the Visualize Results box and clicking the Set As Refit Input button. Within the Refine box, various attributes of the optimization can be changed, including dihedral parameter descriptions and optimization settings. The Run Refitting/Refinement button launches the refinement protocol, which is often substantially faster than the original optimization routine. This is because the initial optimization protocol requires a restrained minimization performed on each target datapoint prior to optimization. The relevant dihedral information is stored indihAll
(seeReference Data
in Visualize Results section), greatly speeding subsequent optimizations/refinements. The user can iteratively adjust dihedral and optimization settings until arriving at an appropriate torsion PES and root-mean-square error (RMSE). - Input/Settings:
- ► Input
- PSF File: The location and filename of the PSF file describing the molecule.
- PDB File: The location and filename of the PDB file containing the QM-optimized molecular geometry.
- Parameter Files: Any parameter files that are required for simulation of the molecule. This includes associated parameter files, as well as the initial parameter file that is under development.
- NAMD binary: The location of the NAMD executable, or simply its name if the location is included in the PATH.
- Output LOG: A location and filename for ffTK output log file (LOG).
- ► QM Target Data
- Location and filenames for Gaussian log files of relaxed potential energy scans.
- ► Dihedral Parameter Settings
- The preferred method for setting the dihedral parameters is via the Read from PAR button, which reads in the missing dihedral parameters from the parameter file under development. The user should modify the periodicity based on knowledge of the type definitions and the bonding geometries and hybridization states of the atoms involved (e.g., for central atoms with sp2 hybridization, n=2). The optimization supports multiplicities, although the user should exercise caution. ffTK only supports phase shift values of 0 and 180 degrees.
- ► Advanced Settings
- Kmax: The maximum value allowed for a force constant.
- Energy Cutoff: The QM energy threshold for inclusion in the optimization. Molecule conformations above this threshold will have their weighting factors set to zero.
- Tolerance: Tolerance to determine the termination criteria use by the optimization function
- Mode: Defines how the optimizer runs; downhill and simulated annealing are supported (see Optimization for details). .
- Write debugging log: If checked, output also includes a .debug.log file that contains detailed information about the setup and calculation.
- Build Run Script: If checked, instead of running the optimization immediately, it generates a script with all of the commands necessary to setup and run the optimization. Useful for running the optimization with VMD in text mode. Note that input files are assigned with absolute paths, and may need to be adjusted if the script is launched from a machine with a different filesystem.
- Output Freq.: The frequency (iteration number) with which data is written to the ffTK output file, and status label updates.
- Save MM Traj.: If checked, the conformations from the MM-relaxed scan will be written as a DCD trajectory to the same directory as the Output LOG.
- ► Visualize Results
- Data Set/RMSE/Plot Color: Attributes for optimization data sets.
- Set Data Color: Change the plot color for selected data sets.
- Plot Selected: Opens a multiplot window and plots selected data sets.
- Include QME/Include MMEi: If checked, includes the QME target data, and the MMEi data in the plot. Default is on.
- Import From LOG: Loads optimization data (initial or refined).
- Write Selected to LOG: Saves data necessary for reloading (via Import from LOG) and optimized parameters for use in BuildPar to update the dihedral parameters for each selected dataset.
- Set As Refit Input: Copies the final parameter data from the selected dataset into the Refine section.
- ► Refine
- The refinement parameters hold equivalent functions to those described in previous sections, but are only applied to the refitting/refinement optimizations.
- Output:
- A ffTK log file for use in updating dihedral parameters in the parameter file under development using BuildPar.
GUI Event Log
- Description:
- Logs the most recent 100 GUI events. The primary functions is to inform the user of GUI actions that lack visual feedback (e.g., update buttons, write buttons).
- Usage:
- Click on GUI Event Log label to turn on/off.


