Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
image size:
246.4KB
Movies of stretching FN-III-1:
real media
(
119.5KB
)
windows media
(
125.9KB
)
mpeg (high quality)
(
2.2MB
)
Image and movie made with VMD
Tissues of the human body are composed of specialized cells held together by a connective fabric of proteins, that form the knots of a net glueing cells together. Upon stretching tissues, the knots unravel, rendering the net larger, but mysteriously also firmer. A protein called fibronectin-III-1 plays a particularly important role in the latter respect. Atomic force microscopy revealed that under mechanical tension fibronectin-III-1 stretches to ten times its initial length; but is does so in two steps, the first stretching step leading to net strengthening. It had been discovered earlier that other fibronectins found between cells are made of two sheets packed like a sandwich, but the structure of fibronectin-III-1 remained elusive. In an experimental-computational collaboration reported recently, the structure has now been resolved that at first sight looked similar to the sandwich structure of the other fibronectins, but on closer inspection showed a weak and a strong sheet. Simulations using NAMD revealed that stretching of the protein unravels readily the weak sheet, and only therafter the strong sheet. It turns out that the strong sheet of fibronectin-III-1 by itself, known as anastellin, inhibits tumor growth. Stretching of fibronectin-III-1, as it occurs naturally in tissue, unravels apparently half of the protein to render it extremely adhesive, strengthening a protein net that prevents metastasis of cancer cells and also assists wound healing (press release, more).
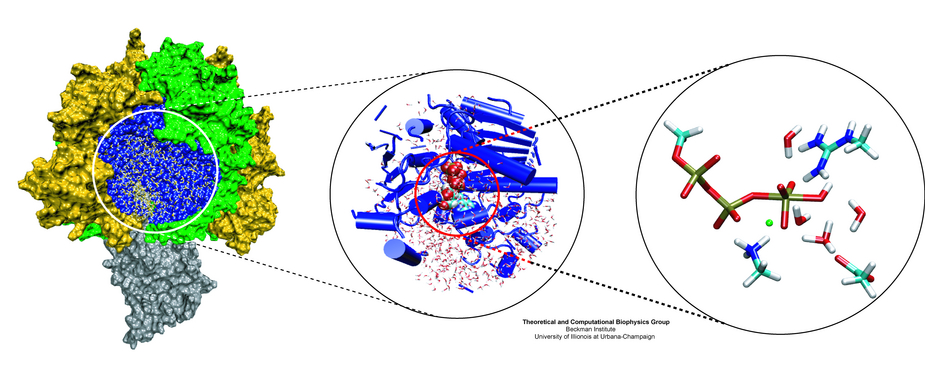
image size:
276.1KB
made with VMD
Adenosine triphosphate (ATP) is the fuel of life; every living cell needs ATP to carry out its functions. The human body actually synthesizes its own weight in ATP every day. This is achieved by ATP synthase, a protein complex of two joined rotating molecular motors. A detailed understanding of how this system functions will impact our fundamental understanding of life, but also the treatment of neurodegenerative diseases or the development of biomimetic nanodevices. Presently, biomedical science still lacks a sufficient understanding of how ATP synthase efficiently stores energy in ATP. A recent report suggests a new chemical mechanism for this energy storage reaction, the discovery being based on quantum-chemical calculations conducted in the presence of the reaction environment in ATP synthase. The suggestion could constitute an important step towards an understanding of one of the most ubiquitous reactions in all living cells, the synthesis of ATP and its use as cellular fuel.

image size:
281.7KB
made with VMD
Our "water
permeation movie"
and the Chemistry Nobel Prize
Winner Science illustration competition
This year's Nobel prize in chemistry goes to Peter Agre and Roderick MacKinnon for their groundbreaking work on membrane channels. We join all in congratulating our two colleagues who have advanced knowledge on membrane channels. Foremost, this advance was made possible through great achievements in experimental methodology by several distinguished researchers, but computational and theoretical investigations of the channels the two new Nobelists investigated, contributed significantly. Our group feels fortunate to have participated in the exciting development through modeling studies of the aquaporin channel (see previous highlights June and April 2003, June and May 2002, and November 2001). Our investigations were based on molecular dynamics simulations of channels in their native membrane environment and demonstrate the widening role of computing in modern life science: using the most advanced computers at the NSF centers in Pittsburgh and Urbana, we could establish the mechanisms of conduction and selectivity in aquaporins in a series of studies that are summarized in our aquaporin web page, and in three recent publications in Biophysical Journal that show how the molecular architecture of channels is optimally designed for their function (1, 2, 3).
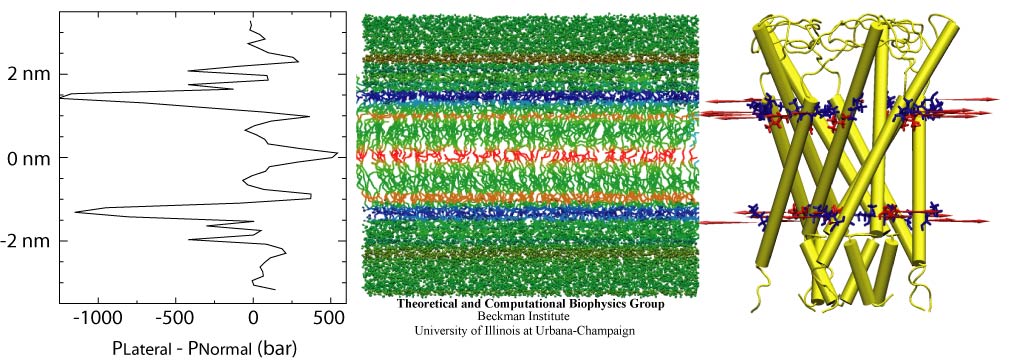
image size: 87.7KB
Living cells sense their environment and respond to its changes through proteins integrated into their outer membrane. These proteins mediate a broad range of cellular activities, including active and passive transport of materials across the membrane as well as response to osmotic shock, which can strain the cell membrane to the point of catastrophic bursting. Cells protect themselves through so-called mechanosensitive channels that open before the membrane tension grows too large. Molecular dynamics simulations and advanced analysis using NAMD and VMD have revealed in a recent report how the joint mechanics of membrane and protein opens a mechanosensitive channel called MscL. The finding promises to revolutionize the modern view of membrane - protein interaction: the membrane, far from being a homogeneous elastic sheet, exhibits a dramatic variation of tension across its thickness that proves to be decisive for the opening of MscL. More here.
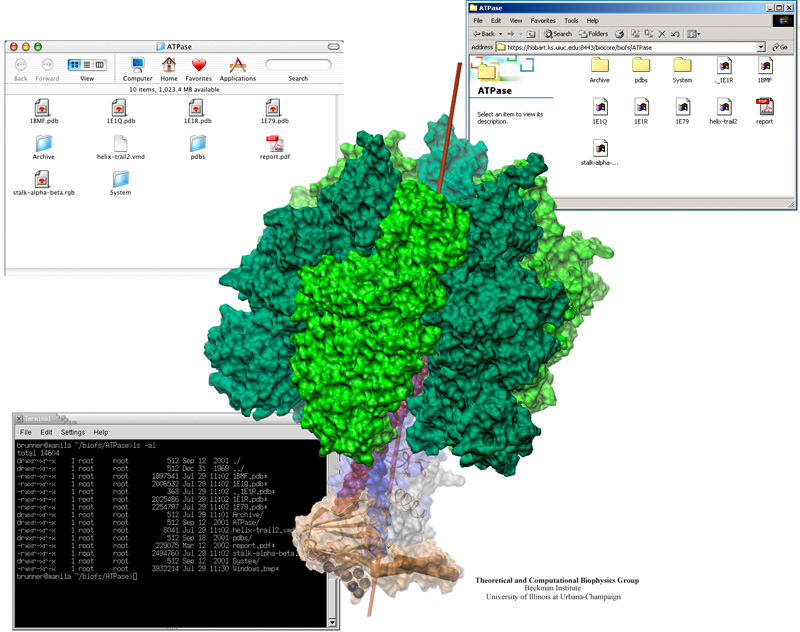
image size: 177.3KB
Biomedical researchers collaborating on a project are often separated by great distances. However, their projects demand continuous sharing of computer files, whether the files be protein data files, trajectories, or written documents. BioCoRE is a web-based collaboration tool for sharing all kinds of data, co-authoring papers, running applications on remote supercomputers, sharing molecular visualization over the Internet, chatting, and keeping an on-line lab book. The newest addition to BioCoRE is the BioFS (BioCoRE File System)-- a way of sharing data that brings ubiquitous computing one step closer to reality. Employing a technology called WebDAV, the BioFS makes files stored centrally within BioCoRE directly available to researchers as if they were stored on their local computer. This provides an intuitive interface, allowing researchers to see folders and files in their native format on their computer desktop. A scientist in Boston can use Word for Windows to edit an upcoming journal article stored in the BioFS. Then a collaborator in Seattle can immediately load the same article on his Macintosh for revision, and another in Germany can complete it on her Linux laptop.
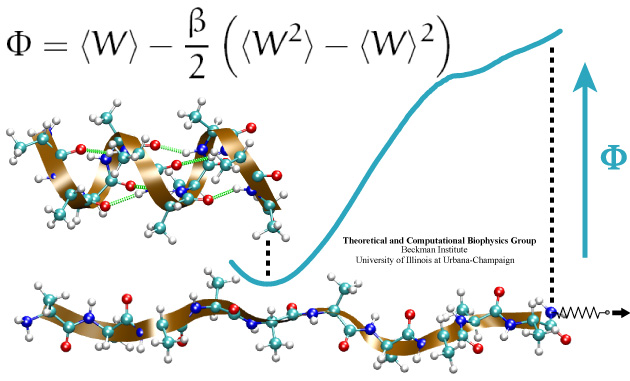
image size:
76.6KB
made with VMD
Many proteins in living cells are nanomachines that undergo mechanical
transformations. Modern modeling methods permit the manipulation of
such proteins to discover the physical mechanism behind their function.
Applying forces, one can induce geometrical changes characteristic of the
proteins' role in the cell and, beyond obtaining qualitative insight,
calculate the work
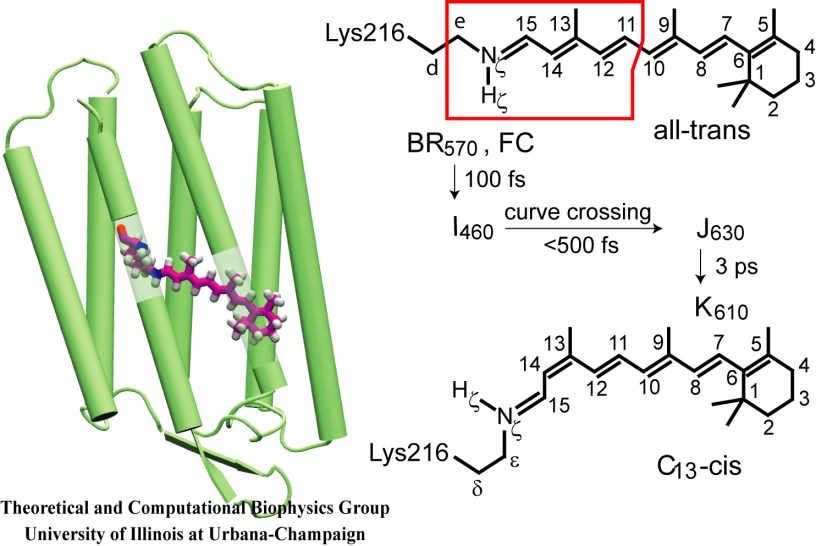
image size:
77.8KB
made with VMD
Retinal proteins are photoreceptors found in many living organisms. They possess a common chromophore, retinal, that upon absorption of light isomerizes and, thereby, triggers biological functions ranging from light energy conversion to phototaxis and vision. The photoisomerization of retinal is extremely fast, highly selective inside the protein matrix, and characterized through optimal sensitivity to incoming light. Photoisomerization takes place while the molecule (retinal) is in its short-lived electronically excited state when internal forces differ strongly from those in the prevalent ground state. Simulation of photoisomerization inside a protein has been a key goal, but unattainable so far since the forces of excited state molecules, largely unknown, need to be described by so-called ab initio quantum mechanical calculations and updated extremely frequently. A recent report describes that this goal has finally been achieved for retinal inside bacteriorhodopsin, a microbial retinal protein that functions as a light-driven proton pump, i.e., a pump driven by photoisomerization and thermal re-isomerization. The description unveils in complete detail the photoisomerization process and the essential role of the protein for the characteristic kinetics and high selectivity of the biomolecular reaction. Supported by comparison with dynamic spectral modulations observed in femtosecond spectroscopy, the results identify the principal molecular motion during photoisomerization that initiate proton pumping. Read more about the project.

image size: 137.8KB
Modeling the molecular processes of biological cells is a craft and an art. Techniques like theoretical and computational skills can be learnt by training, but meaningful applications are achieved only with experience and sensitivity. A summer school in Theoretical and Computational Biophysics attempted to teach both the craft and art of modeling through learning by doing: nearly hundred participants from all over the world came for two weeks to the Beckman Institute in Illinois to stretch proteins, pull water through molecular channels, mine genomic data, build their own computer cluster, and study their favorate biomolecules. After lectures and discussions in the morning, afternoon and evening were devoted to learning by doing, assisted through 300 pages of tutorials, in computer laboratories humming with computational biology software, e.g., VMD, NAMD, and GAMESS, and linked to NCSA's fast pentium machines. The school lasted two weeks, but will go on much longer: all school materials remain available on the web; participants will use BioCoRE to stay in touch and continue the scholarship and friendship experienced in Illinois.

image size:
146.2KB
made with VMD
Biological cells have evolved highly optimized molecular devices made from nanometer size proteins that are the basis for all cellular processes. Engineers seek to conquer the domain of such nanodevices and naturally they look to nature for guidance. In turn, biologists look to nanoengineers for new tools to manipulate cells. A recent study attempts a third type of exchange at the cell - nanotechnology frontier, use of nanodevices that mimic proteins for better understanding of the functions of both: the engineered system can be much simpler than the protein and much easier to comprehend, whereas the protein is more evolved and can reveal the design principles for optimized function. The study links two renowned devices, nanotube and membrane channel. Using the molecular dynamics program NAMD, water flow through nanotubes is simulated and compared with earlier studies of aquaporin water channels in bacteria.
At present, the exchange benefits biology: the very simple nanotube reveals the principles of biologial water conduction, in particular, water flow without loss of electrical voltage. In the future, nanoengineering may benefit, for example, in designing filters for mechanical desalination of sea water.

image size: 284.1KB
Processes in living cells are based on molecular transformations linking reactant and product states. To understand such processes, one needs to know the paths linking reactants and products. Brownian dynamics, as it occurs in cells at physiological temperature, finds any path, as long as it does exist, even if it may take billions of vibrational periods in a protein. But on the nanosecond time scale of computational modeling such paths are too rare to detect, even for the most powerful computers available today. Steered or interactive molecular dynamics , pioneered in our group, can accelerate motion along a reaction path within reach of available computational power, but only if the path is known beforehand. Principles for finding reaction paths are therefore the most desirable goals of theoretical biology. A recent study has formulated a new mathematical principle for determining reaction paths that is based on the mean first-passage time τ linking any intermediate state of the system to the product state. The principle has proven already its worth in describing pathways of light harvesting in photosynthesis, the process that energetically fuels life on earth.
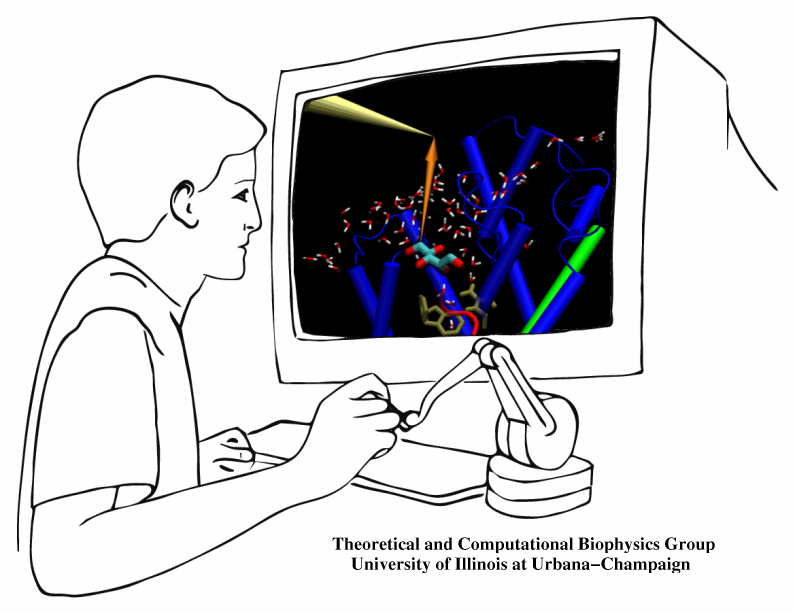
The import of material into biological cells needs to be highly selective. Which physical interactions are the best capable of differentiating suitable from unsuitable imports? How can one keep import traffic fast despite strong selection? As one knows from border crossing, fast traffic and high security can be conflicting objectives. A recent investigation employed a new simulation method to answer the above questions for import of glycerol through a membrane channel. Linking the renown molecular graphics program VMD with the simulation program NAMD permits researchers to view "live" proteins, e.g., the membrane channel, and employing computer game technology, namely, a device that permits one to pull glycerol through the simulated channel, researchers can feel the mechanical resistance of the pulled glycerol, guide it through the channel, and directly observe the selection process involving glycerol's shape, capability for hydrogen bonding, and electric dipoles. The new methodology, interactive molecular dynamics, will revolutionize computational biology and is one of the first tools that take advantage of fast (TeraGrid) computer networks in linking local graphics workstations (running VMD) with a distant supercomputer (running NAMD). The method has received an enthusiastic editorial comment.

image size:
452.7KB
made with VMD
Infections are battled best by the human immune system when there exists a memory from a previous disease or vaccination. The first step in using this line of defense is recognition: Cells of the immune system capture antigens, e.g., microbes in the respiratory tract, then mature in the lymph system, and finally present on their surface pieces of the antigen to T-cells that may recognize the antigen and become activated. The recognition of the antigen by T-cells is dramatically enhanced through surface receptors, CD2 and CD58, on the T-cell and the antigen presenting cell. The receptors stick out from their cell, adhere to one another, and conjoin the T-cell and antigen presenting cell long enough to enable recognition and activation. The molecular basis of this adhesion has been probed in a recent collaborative study with UIUC chemical engineer D. Leckband. Starting from the available crystallographic structure of the CD2-CD58 complex the researchers carried out 90,000 and 100,000 atom simulations using NAMD and pulled the complex apart in steered molecular dynamics simulations. An analysis of the simulations with VMD revealed in atomic level detail how the human immune system is strengthened through elastic adhesion.
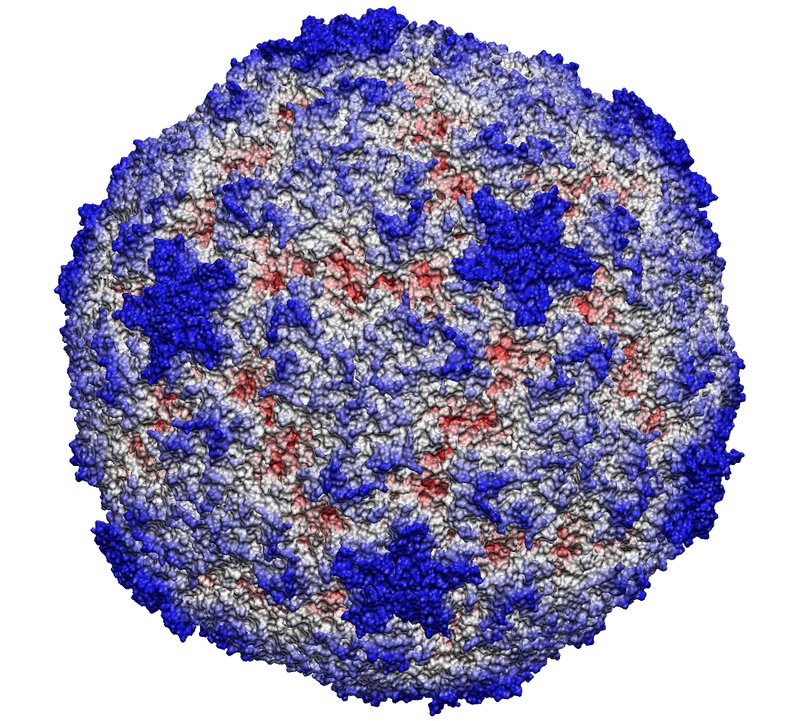
image size:
185.8KB
made with VMD
This month the number of entries in the protein data base passed the 20,000 mark. The data provide microscopic images of the building blocks and functional units of living systems, an invaluable intellectual resource for biomedical researchers. The microscope used is built from software that reads the data base and drives computer graphics, widely available today. Our group's VMD is a leading visualization program for viewing static as well as dynamic structures of proteins and other biomolecules. Now downloaded by more researchers than ever, VMD features ease-of-use for beginners with greater power and flexibility for experts, e.g. as in the case of a 400,000 atom virus capsid. Running on Apple's MacOS X alongside all other common platforms (e.g., Windows, Linux), VMD 1.8.2 offers extensive visualization and analysis capabilities for structures as well as sequences. VMD is easily updated and exceptionally user friendly. VMD integrates closely with its sister programs, the molecular modeling tool NAMD, and the collaboratory suite BioCoRE.
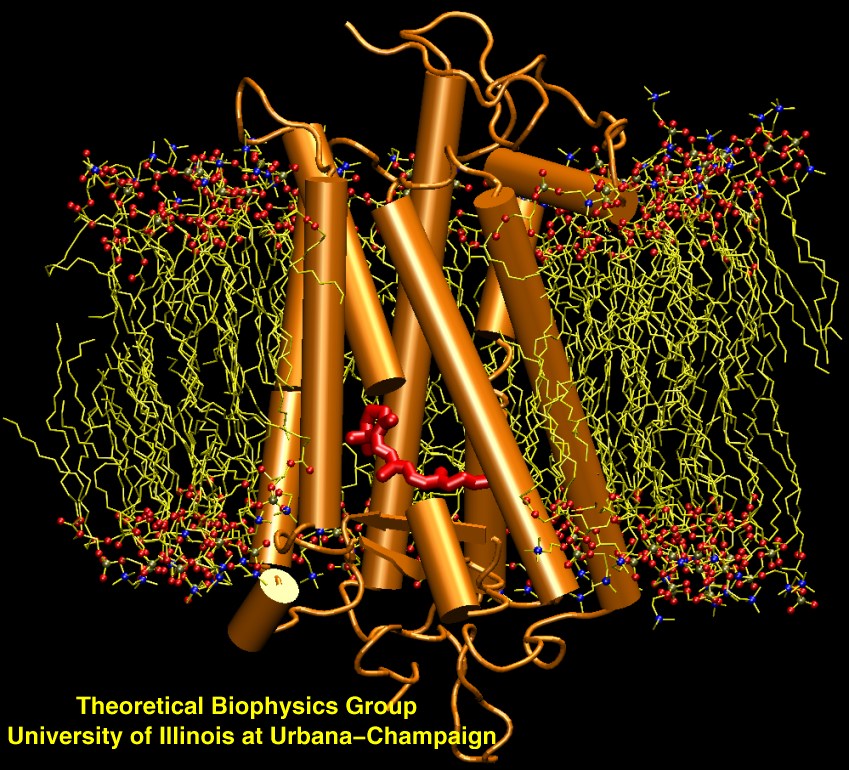
image size:
223.0KB
made with VMD
Scanning their environment for information such as food resources, signs of danger, and illumination is crucial for the well being of biological cells. Evolution has developed for this purpose a great variety of membrane proteins, so-called receptors, that receive physical cues from the external environment through encounters with molecules or absorption of photons and send respective signals into the cell. A common type of receptor sends its signal through interaction with intracellular G-proteins that convey the signal further; proteins of this type are called G protein coupled receptors (GPCRs). GPCRs exist in lower as well as higher life forms and, in fact, the human genome codes for over 1300 GPCRs that detect ions, organic odorants, amines, peptides, proteins, lipids, nucleotides and photons. As about half of modern drugs act on GPCRs, learning how they become activated once they receive their signal is highly relevant. In a recent study, advanced computer simulation techniques using NAMD and VMD have been employed to investigate the first key steps of activation of a GPCR, the visual receptor rhodopsin. more.


