|
Knowledge of the mechanism of association and dissociation of
macromolecules is important for many biological structures and
processes. Among the examples are the binding and dissociation of
substrates of enzyme reactions, the recognition of ligands by their
receptors or of DNA sequences by the DNA binding domains of regulatory
proteins. These processes have in common a transition from one
equilibrium state to another which often is a rare event on the time
scale of molecular dynamics simulations of a few hundreds of
picoseconds. Conventional computational methods for the sampling of
barrier-crossing events increase the probability of unlikely
configurations. Once such configurations have been sampled their
actual occurence can be determined through the known relation between
old and new probabilities. A methodologically related avenue to
characterize rare events through molecular dynamics simulation is the
addition of external forces which reduce the energy barriers. This
approach has the advantage that it corresponds closely to
micromanipulation through atomic force microscopy or optical tweezers.
The external force techniques can be applied to study many
processes, including dissociation of avidin-biotin complex,
dissociation of retinal from bacteriorhodopsin, stretching of DNA,
etc. The molecular dynamics program NAMD,
developed in the group, is capable of performing several different kinds
of SMD, including rotation or translation of one or more atoms. The
group's molecular graphics program VMD
provides a powerful means of visualizing these simulations, and through
the Interactive Molecular Dynamics (IMD) interface can even allow SMD
simulations to be performed in real time.
|
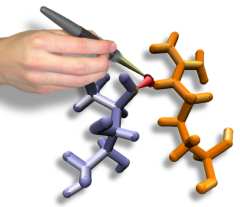
Interactive Molecular Dynamics allows us to pull sugar molecules by
hand through a simulation of the glycerol channel GlpF. As we push
the virtual molecules around, we feel them in our hands as if they
were real. We use this technique to explore features of the channel and
gain new insights into the way it functions. |
 |
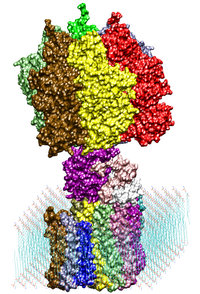 |
Adenosine triphosphate (ATP) is the primary energy "currency" in most
living organisms. ATP synthase is a large (about 100,000 atoms)
protein, which includes a transmembrane F0 unit coupled to
a solvent-exposed F1 unit via a central stalk gamma. The
F0 unit utilizes a transmembrane electrochemical potential
(proton motive force), converting it into the mechanical energy of the
stalk rotation. The rotation leads to cyclic conformational changes in
the catalytic sites in the F1 unit, thereby driving ATP
synthesis. We seek to identify and explore the chain of the elementary
chemical (proton transfer) and mechanical (domain motion) events
involved in the process of converting the electrochemical energy of
the transmembrane proton gradient into the mechanical energy of the
c subunit oligomer rotation. |
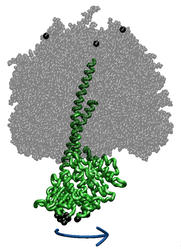 |
ATP synthase is a large multi-protein complex which includes a
transmembrane Fo unit coupled to a solvent-exposed
F1 unit via a central stalk. The stalk rotates within the
surrounding subunits of F1, leading to cyclic
conformational changes in the three catalytic sites in F1
and, thereby, to ATP synthesis. We use steered molecular dynamics to
apply a torque to the central stalk in order to understand the
cooperative interactions that underlie this mechanism. |
| Molecular dynamics simulations induce, over periods of
40 ps to 500 ps, the unbinding of biotin from avidin by means of
external harmonic forces with force constants close to those of AFM
cantilevers. The applied forces are sufficiently large to reduce the
overall binding energy enough to yield unbinding within the
measurement time. |
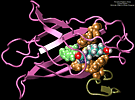 |
 |
We are applying the steered molecular dynamics method to investigate the
action of human synovial protein phospholipase A2 (PLA2) at the lipid water
interface. Our hypothesis is that prior to extruding the phospholipid,
PLA2 must form the tightly bound complex, while the loosely bound complex should
not lead to catalysis.
|
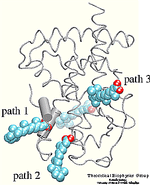
| Formation of bacteriorhodopsin (bR) from the apoprotein and retinal has
been studied experimentally, but the actual pathway, including the
site of retinal entry, is little understood. Molecular dynamics
simulations provide a surprisingly clear prediction. |
 |
 |
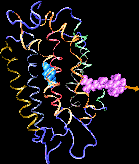
Binding of the hormone to the retinoic acid receptor induces conformational
changes that control and influence gene expression. In order to understand the
functional role of the hormone one must understand the binding mechanism by
which the hormone induces conformational changes. We studied the forced
unbinding of the retinoic acid hormone from its receptor by applying an external
force on the hormone. |
| The giant muscle protein titin, also known as connectin, is a
roughly 30,000 amino acid long filament which plays a number
of important roles in muscle contraction and elasticity. To examine in
atomic detail the dynamics and structure-function relationships of this
behavior, SMD simulations of force-induced titin Ig domain unfolding were
performed. |
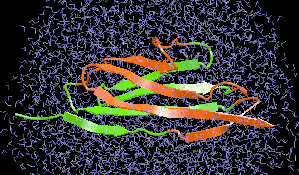 |
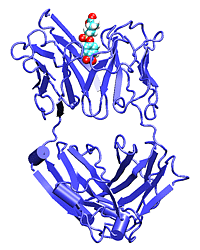 |
Catalytic antibodies are able to bind their antigen and to perform
chemical reaction such as esterification or hydrolysis of the hapten. We
use SMD to unbind the hapten and check the differences in unbinding for
the wild-type and mutant structures.
|
| SMD simulations provide insight into how the bc1 complex performs
its function of separating electrons and protons across a membrane. |
 |
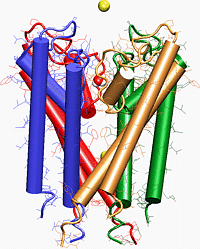 |
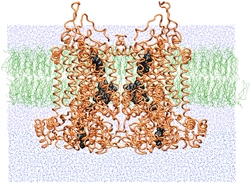
One of the best tools for making the connection between the structure and function of ion channels is molecular dynamics~(MD) simulation, which allows one
to foll ow conformational changes in the structure, and movement of K+ ions across the potassium channel. The difficulty in simulating the passing of K+ or
Na+ ions through the channel can be overcome by using SMD.
|
|


