Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001

Made with VMD
Biological membranes are complex assemblies of diverse lipids,
and molecular dynamics (MD) simulation has been among the most
effective biophysical tools to investigate them at an atomic-detailed level.
Nevertheless, capturing the wide range of lipid compositions and
their mixing continues to pose a major challenge to simulations,
due to the very slow diffusion of lipids.
This issue is particularly important when membrane proteins are present
in the membrane and one is interested in studying how they interact with
lipids. One solution to partially address this problem is to randomize
the initial configuration of lipids in replicate simulations.
The Center has developed a toolkit to rapidly clone a membrane with a
specific lipid composition into as many copies (with shuffled lipids)
as needed within a matter of minutes.
The Membrane Mixer Plugin (MMP)
uses advanced features in
NAMD to remove
clashes and make room before shuffling the lipids;
it also performs several checks to test the quality of the generated membranes.
This method can be accessed via a user-friendly GUI interface in the latest
version of VMD.

Image courtesy L. Casalino. Made with VMD
Members of the Theoretical and Computational Biophysics Group
were part of a talented multi-institutional interdisciplinary team awarded
at Supercomputing 2020 with the internationally recognized
ACM Gordon Bell Special Prize for COVID-19 Research,
described as the "Nobel Prize for high performance computing."
The winning team developed a new AI-driven simulation workflow to
drive 8.5M-atom simulations of the SARS CoV-2 spike protein,
its interaction with the human receptor ACE2,
and 305M-atom simulations of the full SARS CoV-2 virion, using
NAMD on
Summit, the most powerful supercomputer in the United States,
operated by the Oak Ridge National Laboratory.
The NAMD simulations collectively performed over one ZettaFLOP
of calculation.
The AI-driven workflow and weighted ensemble methods used by the
team increased effective performance by orders of magnitude.
VMD was used to
build complete structures, prepare the simulations,
analyze the results, and create all of the team's visualizations
due to its unique capacity for working with
extremely large biomolecular systems and diverse structure data.
Due to decades of prior investment in the development of parallel
molecular dynamics, ongoing technology collaborations with
hardware vendors and national laboratories deploying top tier systems,
and our lab's prior work to enable large all-atom simulations of
viruses and other large systems of up to one-billion atoms, NAMD and VMD
were well-prepared for the challenges posed by the project
and its highly compressed research timeline.
The team set several new high-water marks for the full virion simulation,
molecular dynamics strong scaling performance, the
scale of weighed ensemble accelerated sampling, and the
use of AI-driven simulation for multiscale modeling.
This marks the second Gordon Bell Prize for NAMD, which was
also a winner in 2002.
Read more about the Gordon Bell Special Prize for COVID-19 Research here.
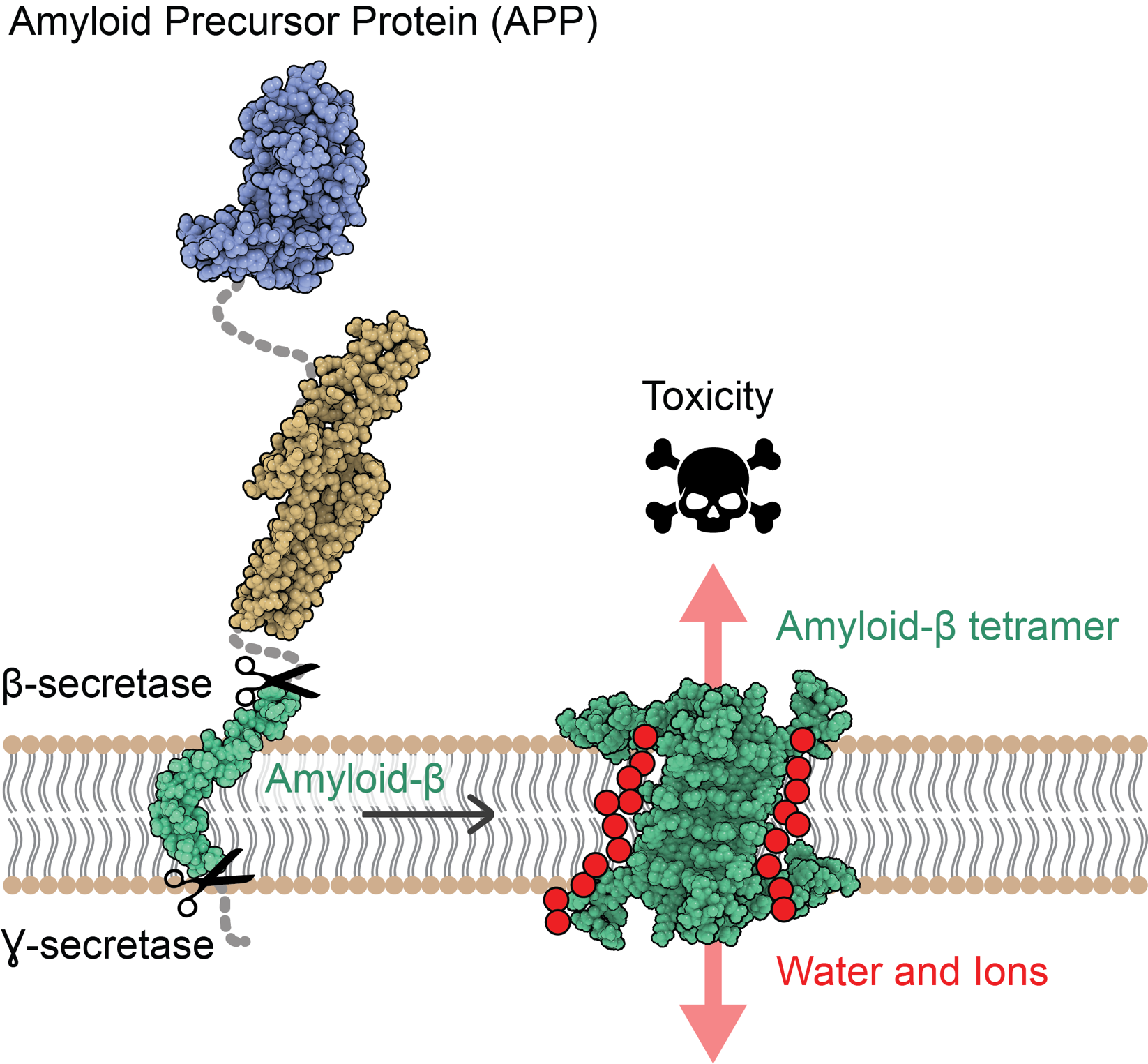
Alzheimer's disease (AD) causes slow and inescapable depletion of neurons in the brain, which leads to the characteristic loss of memory and cognitive function. At a molecular level, AD is caused by aggregation of amyloid-beta (Aβ) protein, however the exact mode of toxicity has remained elusive and highly debated. Inspired by a novel atomic structure of Aβ, and complementary experiments and simulations with NAMD, we report a new mechanism of toxicity for Aβ aggregates. The simulations reveal that Aβ aggregates can perforate the membranes of neurons, compromise their integrity, and consequently trigger their death via the formation of water pores in them. Read more in Nature Communications.

A major goal in molecular simulation, pursued by all programs for decades, is to reach faster simulations and longer timescales to describe, e.g., biological events more meaningfully. The Center has made a major stride in this direction in the recent critical update of NAMD, Version 3.0, which features a fully GPU-resident dynamics scheme for modern GPU architectures and eliminates many of the traditional NAMD overheads, such as its existing communication infrastructures, PCI latency, and the involvement of the CPU during integration, turning NAMD 3 into a completely GPU-bound application. These improvements allow NAMD 3 to perform up to 1.9x faster than v2.13 on existing Volta GPU architectures. It will be up to 1.4x faster on the upcoming NVIDIA A100. Finally, having no involvement of the CPU at all allows NAMD 3 scale perfectly for multi-copy simulation on multi-GPU systems. NAMD 3 performance will grow further as it matures, and as we begin to work on strong scaling with NVLink- and NVSwitch-connected GPUs for single-replica simulations in near future. See more information at most recent NVIDIA developer blog post and in the 2020 NAMD reference paper .
image size:
393.0KB
made with VMD
The cellular membranes in humans, and in any other living cell, are composed of greasy molecules termed "lipids". While originally deemed as a simple medium, we now know that lipids are significant functional regulators for a variety of membrane proteins, including ligand-gated ion channels. As the name suggests, these channels are regulated by binding of specific chemical messengers, e.g., neurotransmitters in the brain, that induce opening of a pore for ions to travel from one side of the membrane to the other. A recent collaborative research between the Center and the group of Claudio Grosman focused on how lipids control the coupling between ligand binding and pore opening. Determining atomic-resolution structures of a ligand-gated ion channel complemented by simulations and free energy calculations performed with NAMD, we show that the response of the channel to ligand binding is completely eliminated in the absence of specific lipids required for activity. Our work highlights how specific protein-lipid interactions are involved in modulating protein functions, and paves the way for further examination of protein activities in complex membrane environments.

Disease due to a viral or bacterial infection continues to pose a major health concern, particularly today in the midst of the COVID-19 pandemic. Fighting such health threats requires the development of new antiviral and antibiotic drugs, a process that is increasingly guided by molecular modeling and computer simulation. The recently released NAMD 2.14 is now especially well suited to handle very large molecular systems for cell-scale simulations and to take advantage of massively parallel, GPU-accelerated architectures like the Summit supercomputer at Oak Ridge National Laboratory, currently ranked as the fastest in the world. Recent Summit simulations of a one-billion-atom protocell system, assembled to emulate a minimal but realistic cellular envelope and shown here in a cross-sectional view, push the limits of constructing and modeling atomic-resolution biological structures, paving the way for improved methodologies and tools that will enable the simulation of large viral and bacterial complexes.

For too long, cellular membranes were considered to merely provide a defense wall for the cell. However, we now know that they constitute one of the most fundamental compartments for the function of a living cell. Key processes such as selective transport and exchange of information between the cell and its environment are mediated by membrane proteins, key players that also form a major site of engagement both for pathogenic species such as viruses to enter the cell and for many drugs to bind to and to exert their beneficial effects. Fascinatingly, we are learning that lipids (fat molecules constituting the cellular membrane) are not just sitting there; rather, they can specifically regulate the function of membrane proteins through diverse mechanisms. But it is very hard to "see" how they accomplish this task. Using microscopic simulations with NAMD and in close concert with advanced experimental techniques, the Center has investigated a broad range of such functional lipid-protein interactions in several recent studies [1,2,3,4,5,6,7], not only identifying specific binding sites for signaling lipids (e. g., cholesterol, or charged lipids such as PIP2 and PIP3) on membrane proteins (e. g., transporters, channels, receptors) with key physiological roles, but also providing important information on how the function of these proteins can be regulated by lipids. These studies have greatly enriched our knowledge on the regulatory roles of lipids in membrane biology and protein function.


