The p7 viroporin
HCV
Hepatitis C virus (HCV) is a small enveloped RNA virus of the family Flaviviridae, which is a leading cause of chronic hepatitis, liver cirrhosis and hepatocellular carcinoma, and affects over 2% of the world's population. Current therapies based on pegylated interferon and ribavirin are poorly tolerated by patients and ineffective in up to 50% of cases, making the identification of new drug targets a priority.
The HCV genome consists of a 9.6-kb positive-stranded RNA molecule encoding a single polyprotein precursor, later processed by both host and viral proteases into ten separate proteins. These are: the Core protein, which composes the nucleocapsid; the viral envelope E1 and E2 glycoproteins; the p7 viroporin; NS2, required for virus assembly; and the replication machinery consisting of NS3, NS4A, NS4B, NS5A, and NS5B (reviewed in Moradpour et al.).
p7
The p7 protein has recently attracted interest as a possible new drug target. For example, the small molecule BIT225, which is believed to inhibit the ion-channel activity of p7, has shown promising results in recent clinical trials, though its mechanism of action is not yet well understood.
The ability of p7 to control the permeability of the membrane to ions and to facilitate virus production qualifies it as a viroporin, alongside with, for instance, M2 from influenza A virus and Vpu from HIV-1. The essential features of a viroporin are that it forms small membrane-spanning structural units, usually consisting of one or two helices that can oligomerize into a channel. The resulting structures range in complexity from passive, non-selective pores, which allow ions to flow through in an uncontrolled fashion, to selective ion channels with a specific gating mechanism, as found, for example, in M2. Weakly selective ion channels like Vpu fall somewhere in the middle, exhibiting "channel-pore dualism".
Modeling p7
An atomic model of the p7 monomer (genotype 1b, strain HCV-J, accession number D90208) was determined prevsiously via NMR and MD by Montserret et al. This monomeric model was used to construct oligomeric models with 4 to 7 subunits by applying the appropriate symmetry operations. In general, two types of oligomeric states were constructed, based on models previously proposed in the literature - one in which the monomers extend radially away from the center and have minimal contact with one another, similar to the model described in Patargias et al. , and one in which the monomers are rotated such that adjacent subunits make better contact, as in to the models described in Clarke et al., and StGelais et al.. Heptameric and hexameric models were constructed of the first type (Heptamer A, Hexamer A) , and heptameric, hexameric, pentameric, and tetrameric models of the second type (Heptamer B, Hexamer B, Pentamer, Tetramer) were constructed. Additionally, a third hexameric model was constructed which had the opposite handedness compared to the rest of the models (Hexamer C). All models are shown in Figure 1.
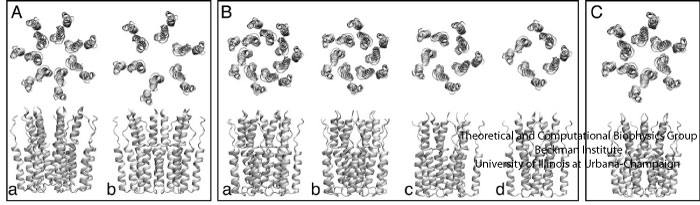
Fig. 1. Oligomeric models constructed from the p7 monomer. Box A: (a) Heptamer A, (b) Hexamer A; Box B: (a) Heptamer B, (b) Hexamer B, (c) pentamer, (d) tetramer; Box C: Hexamer C. The models in box A were constructed so that the monomers extend radially away from the center, similar to the model presented in Patargias et al.. In contrast, the models in box B were built such that the monomers optimize the contacts between their mutual surfaces, consistent with the models described in Clarke et al., and StGelais et al.. The model in box C was built to be similar to, but have the opposite handedness of Hexamer B.
All of the models, with the exception of Hexamer A, were structurally stable over the course of an equilibrium MD simulation of approximately 100 ns. Since experimental results suggest that p7 primarily exists as some combination of hexamers and heptamers in vivo, it was expected that the Hexamer B and Heptamer B structures would be favorable. The tetrameric and pentameric models also turned out to be quite stable in simulation. p7 has yet to be observed in vitro as a tetramer or pentamer, but it is possible that such structures may be viable, possibly as intermediate stages in p7 self-assembly prior to the formation of the final hexameric or heptameric complexes. The Hexamer A model collapsed within a few nanoseconds of simulation time. The Heptamer A model did not collapse, but instead rearranged itself slightly in order to have better inter-subunit contacts.
The Central Pore
The majority of the p7 models (Heptamer B, Heptamer A, pentamer, tetramer) formed a continuously open solvent-accessible pore. Hexamer A collapsed early in the simulation, Hexamer B was initially sealed but opened spontaneously over the course of the simulation, and Hexamer C had to be induced to open. In the case of Hexamer B, the pore was initally blocked in two places by rings of adjacent hydrophobic residue sidechains. One seal was at the level of F25, already proposed to play a role in gating the channel, with the second at the level of I32, whose purpose and position in the channel has been largely unexplored. The F25 seal can be seen in the pore-radius profile of Hexamer B (see Figure 2), which depicts the width of the pore along the longitudinal axis of the complex. In the first 65 ns of the simulation, the protrusion of the F25 ring constricts the pore to a radius smaller than that of a water molecule (solid line and red line, respectively, Figure 2). The barrier formed by the F25 side chains recedes when the pore opens, allowing water permeation (dotted line, Figure 2). By contrast, the pore-radius profiles of the rest of models show no constrictions narrow enough to preclude water diffusion (Figure 2). The I32 side chains are not bulky enough to block the channel; possibly they prevent the passage of water by withholding hydrogen-bonding partners. In the Hexamer B simulation, the channel opened spontaneously due to the interaction of water molecules with the F25 side chains (movie of Hexamer B trajectory). The Hexamer C model was also initially sealed, this time at the level of Y31 and L20. We did not observe spontaneous opening of the central pore during the simulation. In order to induce the pore to open, a brief electric field was applied in the simulation, which reoriented the side chains of the pore-lining residues enough to open the pore. The Hexamer C model then remained in the open conformation for the remainder of the equilibrium simulation.
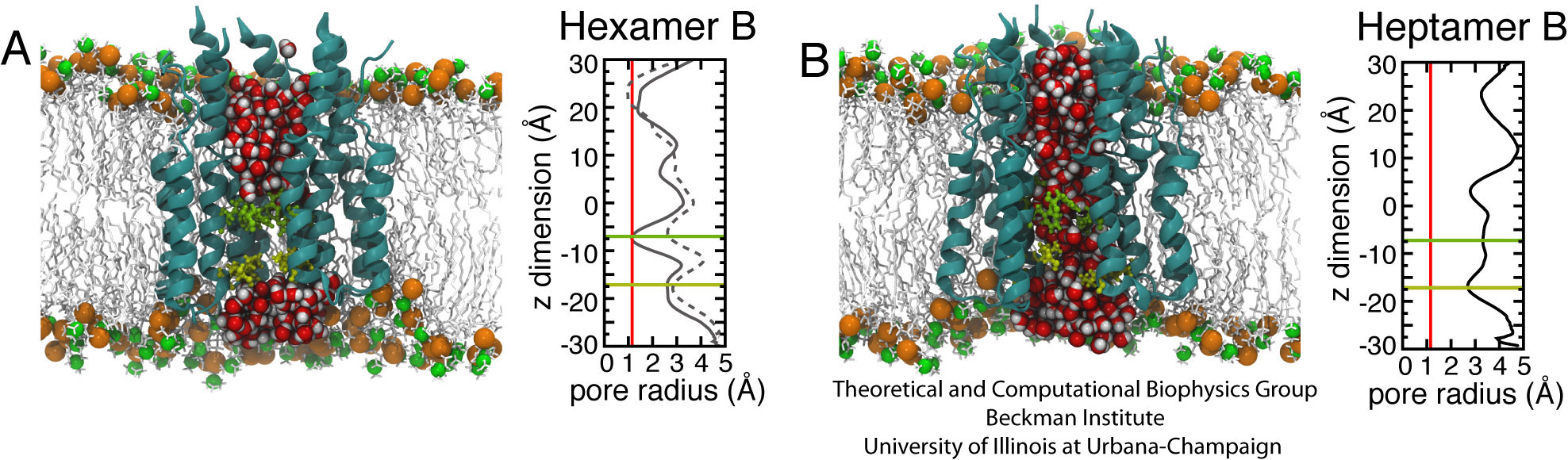
Fig. 2. Molecular assemblies and pore-radius profiles of p7 Hexamer B (A) and Heptamer B (B) pores in a POPC bilayer. Left panels: side views of Hexamer B and Heptamer B (ribbon representation) with water molecules inside the central pore represented as van der Waals spheres (red and grey). The side chains of F25 (in green) and I32 (in yellow) are represented as sticks. The model POPC bilayer is described by means of a stick representation (light grey), with phosphorus and nitrogen atoms displayed as glossy orange and green van der Waals spheres, respectively. Right panels: radial profiles depicting the width of the pore along the longitudinal axis of the complex were calculated using the program HOLE. The red line is positioned at 1.4 A, the approximate radius of a water molecule. The green and yellow lines mark the approximate positions of F25 and I32, respectively. In the Hexamer B plot (A), the solid line represents the radial profile before the opening of the pore and highlights a constriction at the level of F25. The dotted line denotes the radial profile after the pore has opened (the protrusion near z = 25 A is the result of association between the N-terminal tails of the six subunits in the solvent below the membrane, and does not represent a blockage of the pore).
Most of the p7 oligomer models were constructed to be right-handed as viewed from the terminal side. This was considered to be the most promising arrangement, in terms of placing favorable residues towards the center of the pore. In particular, H17 and F25 face the pore in this configuration, as has already been proposed, but also the hydrophilic S21. The Hexamer C model was constructed with the opposite handedness, to see how such a model would fare during simulation. However, Hexamer C has mostly hydrophobic residues facing the pore, which seems like an unfavorable situation for an ion channel. Additionally, the arrangement of the monomers in Hexamer C left H17 facing the body of the protein, rather than the pore, though over the course of the simulation, some of the subunits rotated slightly, giving H17 minimal access to the central pore. Interestingly, the pore-lining residues of Heptamer A are similar to those of Hexamer C, and over the course of the simulation, it rearranged itself slightly to have better inter-subunit contact, eventaully settling into a conformation with a slight left-handed bias. Our simulations thus cannot rule out the possibility of left-handed p7 oligomers; however, the set of pore-lining residues seems inconsistent with existing data.
Reconciling with EM density
In cryo-EM experiments, Luik et al. observed hexameric p7 complexes in a highly tilted flower-like conformation, in contrast to the upright p7 oligomer models considered so far. However, the p7 complexes in the experiments of Luik et al. were formed in very short DHPC lipids, which form a much thinner membrane than the POPC lipids used in the simulations (and as would be found in the endoplasmic reticulum, where p7 is thought to assemble in the cell). Using an EM density map provided by Luik et al., a tilted p7 hexamer model was constructed by using Molecular Dynamics Flexible Fitting (MDFF) to drive the upright Hexamer B model into the EM data (movie of MDFF fitting). This model was then placed in both DHPC and POPC lipids and allowed to equilibrate. In the DHPC membrane, the p7 model largely retained its bent shape, whereas in the POPC lipids, hydrophobic mismatch forced the p7 model to begin to straighten up back towards its original upright conformation. Both conformations are depicted in Figure 3. These simulations suggest that p7 is capable of flexibly adapting to the external lipid environment, and that it is reasonable to expect to find p7 in a different conformation in short lipids like DHPC vs. ER-like POPC lipids. Changes in the lipid environemnt may even influence the gating of the channel, as has been shown with other viroporins such as Vpu from HIV.
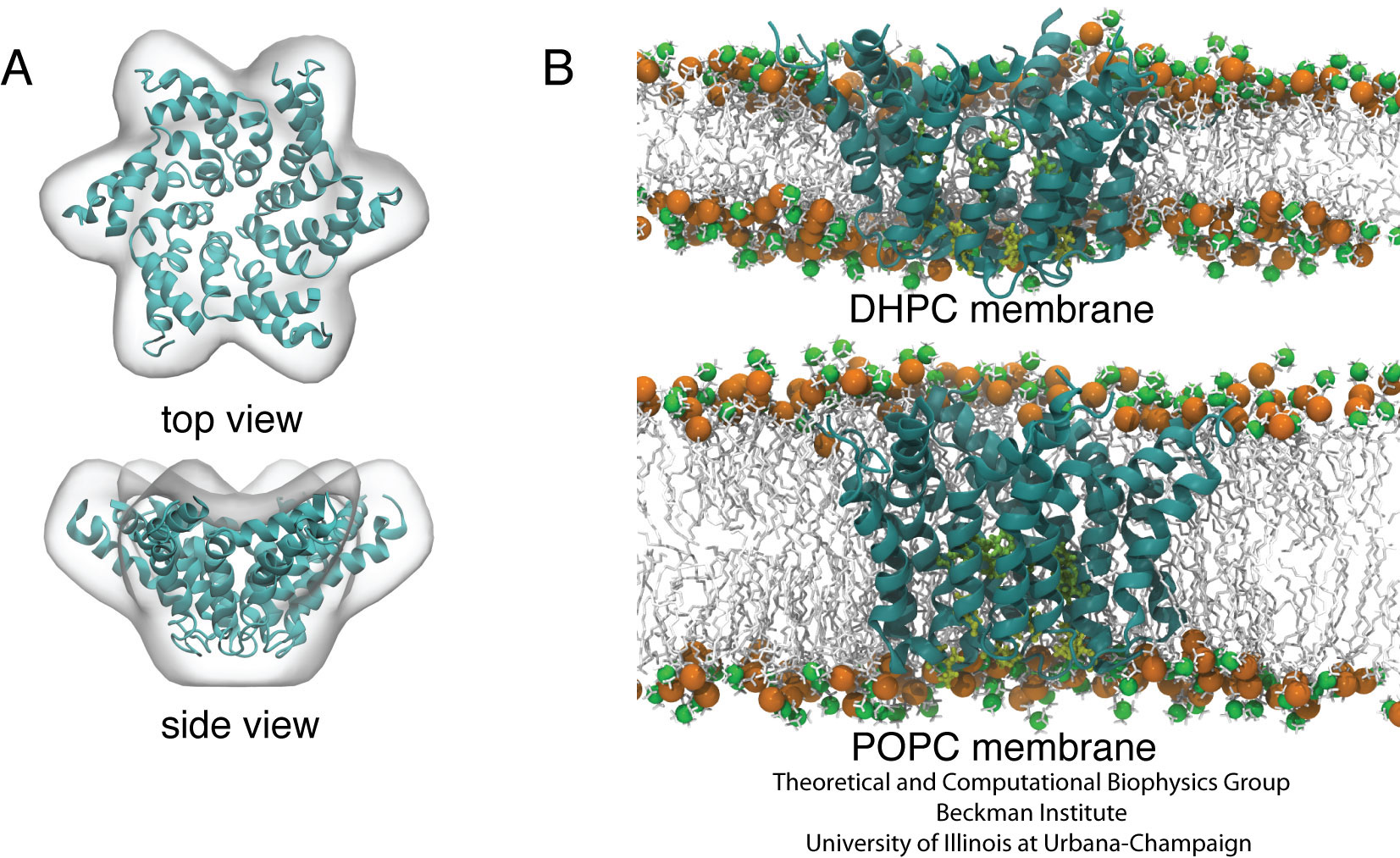
Fig. 3. Conformation of the p7 hexamer in short- and long- tail lipid environments. (A) Hexamer B model (ribbon representation) fitted into the EM envelope of Luik et al. (B) Side views of the EM-fitted hexamer model in the DHPC and the POPC lipid bilayers.
TCB Publications
The p7 Protein of Hepatitis C Virus Forms Structurally Plastic, Minimalist Ion Channels. D. E. Chandler, F. Penin, K. Schulten, and C. Chipot. PLoS Computational Biology, 8(9), 2012.
Investigators
maintained by Danielle Chandler


