26S Proteasome - The Protein Recycling System of the Cell

|
|
|
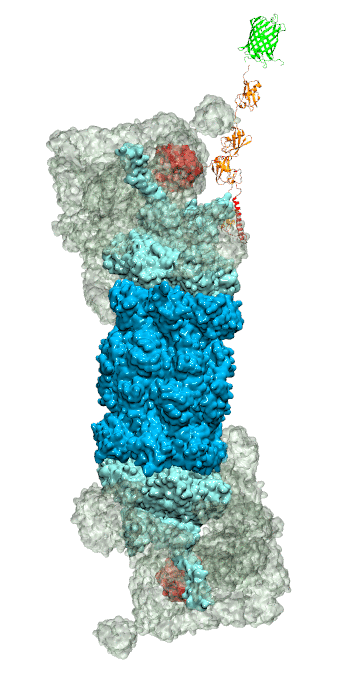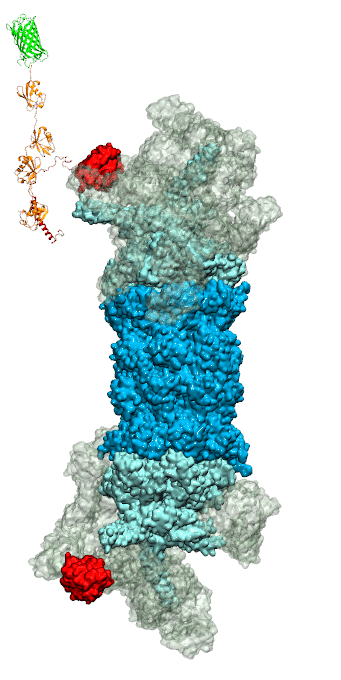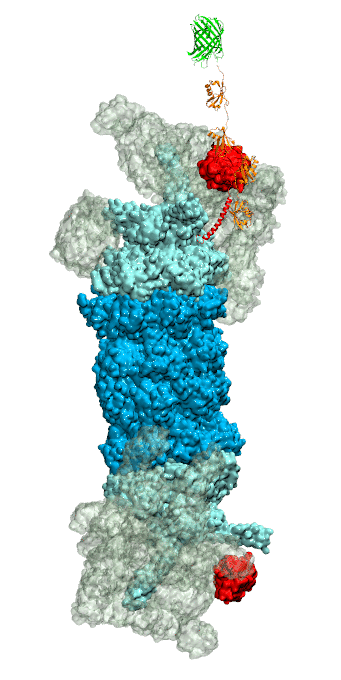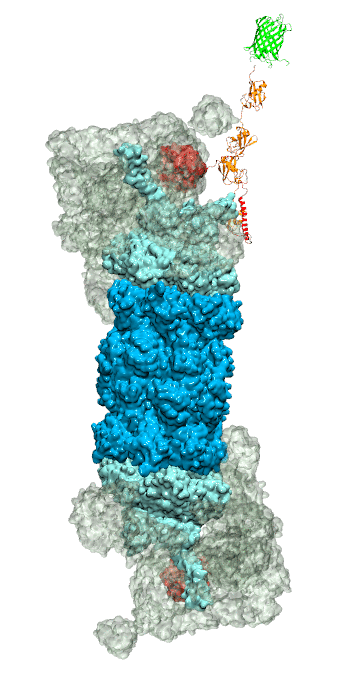
Figure 1 - The recycling system of the cell. The 26S proteasome is the key player of the human protein recycling system. The first structure of the human 26S proteasome obtained through integrative modeling will lead to breakthroughs in understanding its detailed function and will play a pivotal role in the development of the 26S proteasome as a drug target for molecular disease therapies. |
|
|
Overview
While waste recycling became popular in our daily life more recently, living cells have mastered recycling of their protein content since their very beginning. Recycling of unneeded protein molecules in cells is performed by a molecular machine called 26S proteasome (Figure 1), which cuts these proteins into smaller pieces for reuse as building blocks for new proteins. Proteins that need to be recycled are labeled by tags made of poly-ubiquitin protein chains. The 26S proteasome machine recognizes and binds to these tags, pulls the tagged protein close, then unwinds it, and finally cuts it into pieces.
As the cell's recycling machinery, the 26S proteasome is vital for a variety of essential cellular processes, including protein quality control, cell cycle regulation, adaptive immune response, and apoptosis. The 26S proteasome is responsible for the vast majority of regulated intracellular protein degradation and is an important drug target for multiple diseases, including cancer, neurodegenerative diseases, and immuno-inflammatory disorders. The 26S proteasome recruits, unfolds, and degrades poly-ubiquitin tagged proteins through a complex interaction clockwork of over 60 known protein subunits that is driven through ATP hydrolysis.
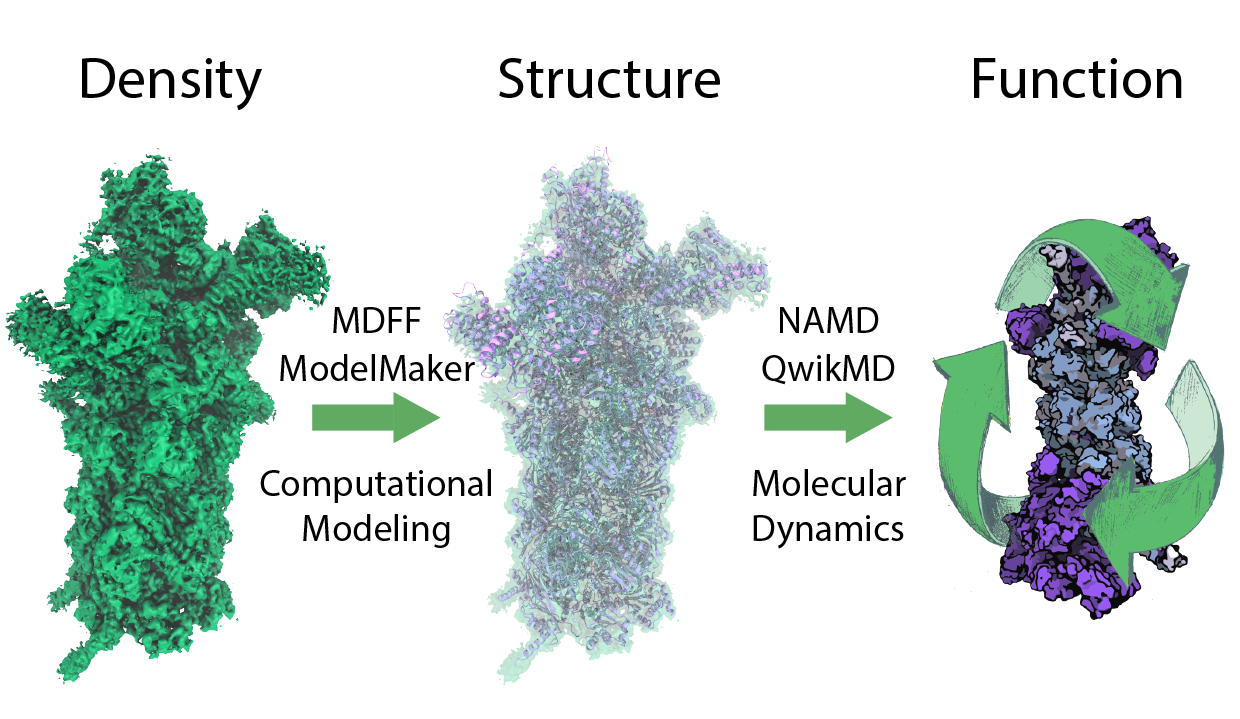
Despite its substantial role in the cell's life cycle, the proteasome's atomic structure and function remained elusive. However, recent developments in hybrid experimental methods based on the revolutionary advance of electron microscopy together with improvements in real space refinement methods have led to previously unimaginable structural information on large macromolecular complexes . The resulting structures are used as initial structures for molecular dynamics simulation to investigate the dynamics underlying protein function (Figure 2). For readers who want to start with density guided modeling we recommend the MDFF tutorial for QwikMD and for more advanced users the MDFF and ModelMaker tutorials.
Atomic Structure of the 26S Proteasome
|
|
|
Figure 2 - Understanding protein function. In order to understand protein function at atomic detail, structure and dynamics need to be connected to function. Molecular dynamics simulations are well suited for this task, however initial atomic structures are needed. To derive these initial structures from experimental data, like X-ray or cryo-EM densities, integrative modeling combining molecular dynamics flexible fitting (MDFF) with de novo structure prediction algorithms and Mote Carlo rotamer search are employed. |
We obtained the first atomic structure of the human 26S proteasome with bound nucleotides based on a 3.9 Å resolution cryo-EM density obtained by integrative modeling combining molecular dynamics flexible fitting (MDFF) with de novo structure prediction algorithms. Furthermore, utilizing molecular dynamics (MD), steered molecular dynamics (SMD) simulations, and generalized simulated annealing (GSA) techniques we revealed the underlying mechanism of poly-ubiquitin tagged protein recognition and recruitment through the 26S proteasome .
To build an accurate atomic model of the human 26S proteasome we followed the strategy established for large macromolecular complexes. We first built comparative models of the human 26S proteasome subunits based on the yeast 26S proteasome and extended structurally unresolved segments by de novo modeling. Next, we refined the structure according to the density in real space by combining MDFF with de novo structure prediction as well as Monte Carlo-based backbone and side chain rotamer search algorithms in an iterative manner (Figure 2). The features observed in the resulting structure are important for coordinating the proteasomal subunits during substrate processing. One of the key novel features is that in the predominant state of the heterohexameric ATPase motor of the proteasome there are one ADP and five ATP bound. The structure of the ADP bound subunit is distinct from the other five subunits most notably in the pore loop region, which is known to be a key region interacting with the substrate during unfolding.
Substrate Recognition by the 26S Proteasome
|
|
|
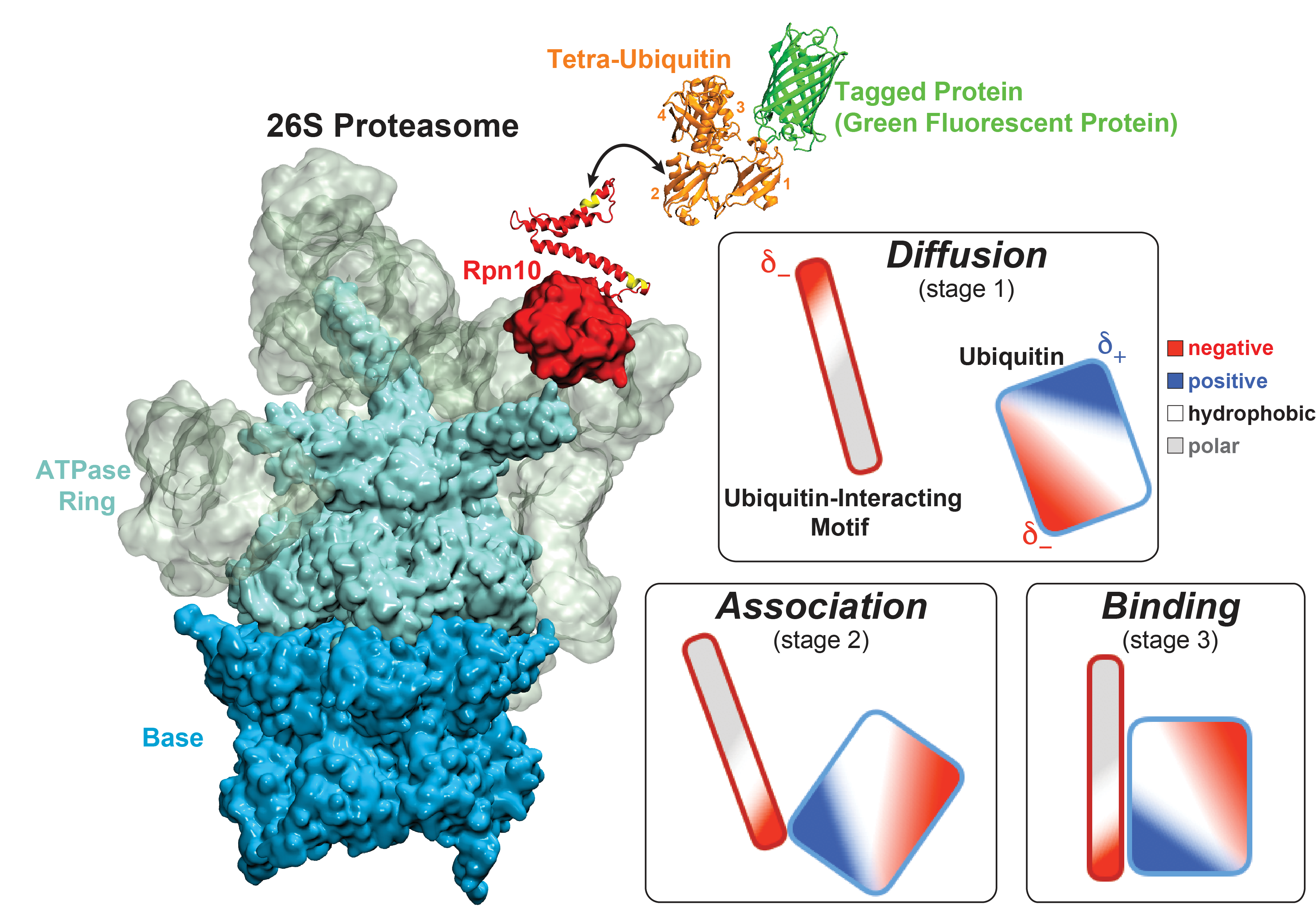
Figure 3 - Recognition of the tetra-ubiquitin tag by the 26S proteasome in three stages: In stage 1 of the recognition process conserved complementary electrostatic patterns of Rpn10 and ubiquitins guide protein association; stage 2 induces refolding of Rpn10 and tetra-ubiquitin tag; stage 3 facilitates formation of hydrophobic contacts between the tag and Rpn10. |
Furthermore, we employed MD, sMD and GSA simulations, utilizing QwikMD, our new intuitive "point and click" graphical interface connecting VMD and NAMD that significantly simplifies the setup, execution, and analysis of NAMD simulations ranging from laptop computers up to supercomputers. Our simulations elucidated the process of how ubiquitin-tagged proteins are recognized by the partially disordered flexible arm of the ubiquitin receptor Rpn10 of the 26S proteasome (Figure 3). Rpn10 consists of a globular domain that binds to the 26S proteasome and a flexible arm that contains ubiquitin-interacting motifs (UIM). Previous studies identified that ubiquitin binds the UIMs through hydrophobic interactions, however, poly-ubiquitin mostly assumes closed forms in solution, in which the hydrophobic patches are protected. Our results reveal that hydrophobic UIMs of Rpn10, which bind to hydrophobic patches of ubiquitin, are likewise mostly protected prior to binding. Therefore, refolding of both tetra-ubiquitin and Rpn10 to reveal hydrophobic patches is a necessary step of the binding process. Since it is unlikely that protected hydrophobic patches can initiate refolding and efficiently search for each other, our results indicate electrostatic interactions to be responsible for the initial stage of the mutual recognition between UIMs and Rpn10.
All in all, the combination of MDFF with Monte Carlo-based rotamer search algorithms enabled us to obtain a structural model based on high-resolution (< 4.0 Å) cryo-EM densities and obtain new insights on substrate recognition and unfolding. The obtained atomic structure permit further computational studies of proteasomal function and will serve as a starting point for future structure-guided drug discovery which will assist in developing the proteasome as a drug target.
Related Highlights
Overcoming the Challenges of High-Resolution Data
QwikMD - Gateway to Easy Simulation


