A prototype molecular motor stepping on ssDNA: PcrA helicase
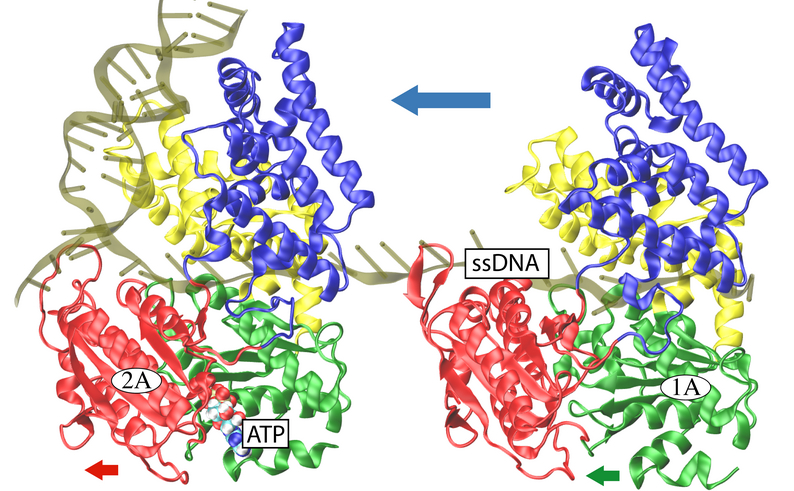
DNA helicases are ATP driven molecular motors involved in all aspects of DNA metabolism, including transcription, replication and recombination. They catalyze the separation of double stranded DNA (dsDNA) into its constituent single stranded DNA (ssDNA) components. Monomeric PcrA helicase from Bacillus stearothermophilus is one of the smallest molecular motors known to date with full atomic scale structures available. The DNA unwinding action of PcrA is directly caused by helicase translocation along ssDNA, which is driven by the hydrolysis of ATP in a single catalytic site. The translocation proceeds unidirectionally from 3' to 5' end of ssDNA, at a rate about 50 nucleotides per second; that is to say, it would take about tens of milliseconds for PcrA to move one nucleotide distance on the ssDNA. Previously, an inchworm model was proposed for the translocation, in which PcrA moves by alternating affinities between domains (1A and 2A) and ssDNA, although the physical basis of the model was not established.
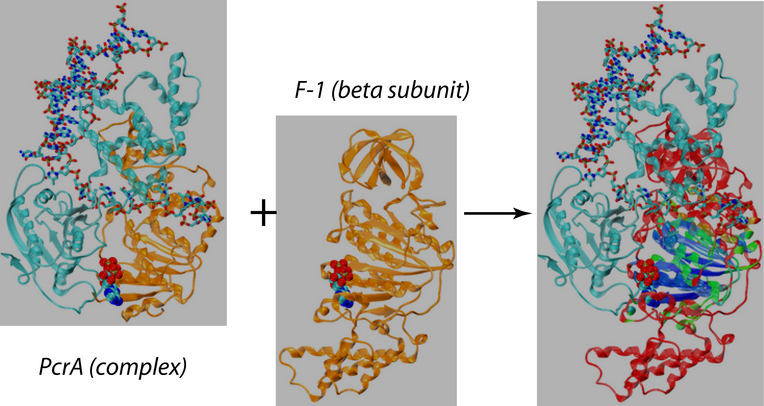
Belonging to helicase superfamily I (SFI, which also includes Rep and UvrD helicases that are structurally homologous to PcrA), and similar to the ATPases associated with diverse cellular activities (AAA+) family, PcrA helicase has two RecA -like domains 1A (green) and 2A (red) (see figure on the left and a linked image) which are proposed to function as the translocase; a strand of ssDNA threads through the protein above the domains 2A and 1A while ATP binds into a cleft between these two domains. The ATP binding site is lined by amino acids that are highly conserved among SF1 heliases; two of the motifs in the ATP binding site (Walker A and B) are highly conserved among all ATPases. Indeed, superimposing the ATP binding pocket of PcrA helicase with that of F1-ATPase shows that the sites have high structural identities (see the blue domain region in the figure right below), suggesting a commmon mechanism of ATP hydrolysis.
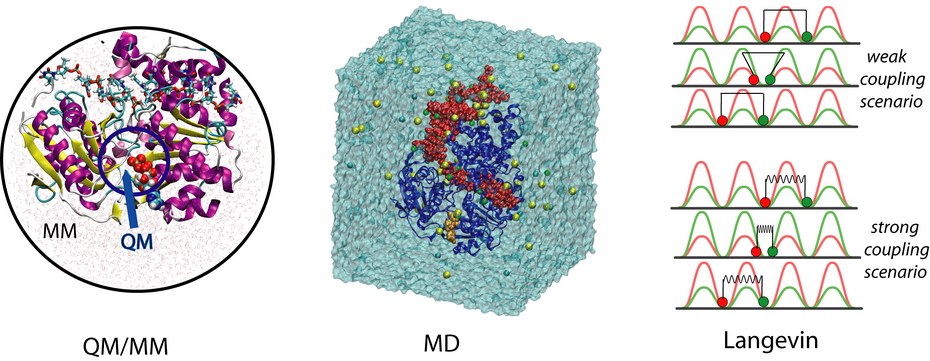
Although functionally amazing, molecular motor proteins present formidable challenges to the computational researcher. Typically, the operation cycle of a molecular motor spans a wide range of length and time scales, from the localized (about 1 angstrom) and fast chemical reaction (about femtoseconds) in the catalytic site(s) to the large scale (up to 10 nanometers) and much slower conformational motions (up to milliseconds) involved in the motors' physiological function. From a computational point of view, this means that currently there exists no single approach capable of capturing the whole spectrum of events during molecular motor function. To overcome this hurdle, we combine three comutational approaches, quantum mechanical/molecular mechanical (QM/MM) calculation, classical molecular dynamics (MD) simulation and stochastic modelling together, in order to study this prototype molecular motor, PcrA helicase, from the electronic to the functional level (read a recent review ). These approaches are illustrated below.
QM/MM studies on ATP hydrolysis and force generation
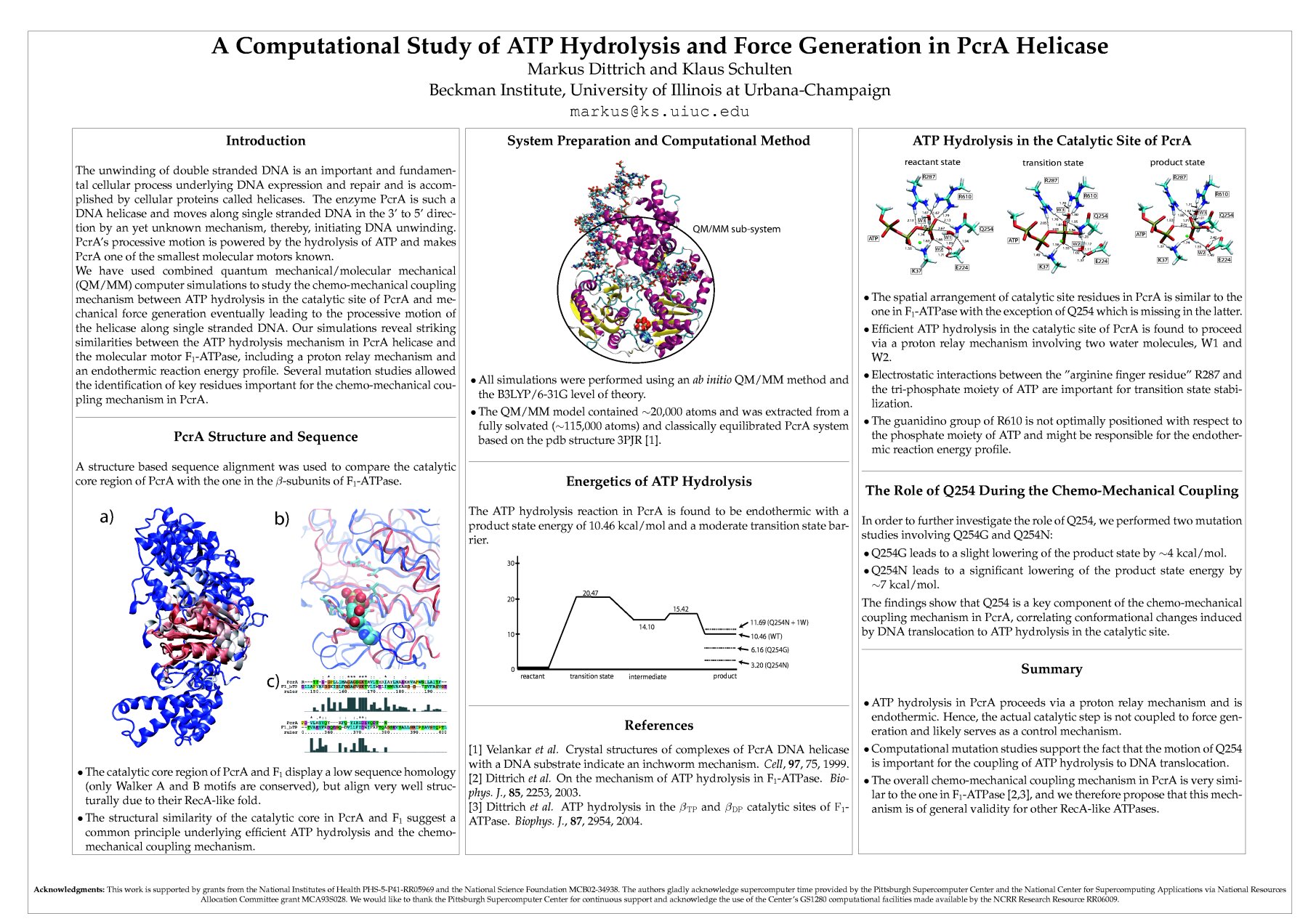
At the catalytic site level, combined QM/MM simulations were used to study the chemo-mechanical coupling mechanism in PcrA, i.e. the means by which ATP hydrolysis in the catalytic site is coupled to protein conformational changes which eventually give rise to translocation along DNA. QM/MM simulations allow one to study quantum mechanically the chemical reaction (ATP hydrolysis) inside the catalytic binding pocket and the large biomolecular environment provided by PcrA which is simulated using classical molecular mechanics methods. The PcrA model used for our studies, consisted of about 20,000 atoms, 77 of which were treated quantum mechanically at the B3LYP/6-31G level of theory (see a recent publication ).
A structure based sequence alignment of the catalytic site of PcrA with the ones of F1-ATPase revealed their striking similarity suggesting a common mechanism by which the chemo-mechanical coupling is achieved. Our QM/MM simulations provide several important new insights into ATP hydrolysis coupled DNA translocation in PcrA. Similar to our previous studies of the chemo-mechanical coupling in F1-ATPase (Dittrich, 2003 and 2004 ), a proton relay mechanism was identified as the physiologically relevant proton transfer pathway mediating nucleophilic attack, providing further evidence for its catalytic importance in RecA-like ATPases. The "arginine finger" residue R287 reaching into the catalytic site from a neighboring protein subunit was found to be crucial for transition state stabilization, thereby, providing a means of coupling large scale protein conformational changes to the catalytic reaction and adding to the list of similarities between PcrA and F1-ATPase at the catalytic site level. Employing computational mutation studies, we were able to show that the position of the glutamine residue Q254 with respect to the ATP bound in the catalytic site greatly influences the energetics of the ATP hydrolysis reaction. Since Q254 is part of a loop region involved in DNA translocation through PcrA its influence on the ATP hydrolysis reaction suggests a means by which protein conformational changes induced by DNA translocation are coupled to the catalytic reaction in the binding site of PcrA and vice versa.
MD/SMD simulation on molecular basis of the translocation
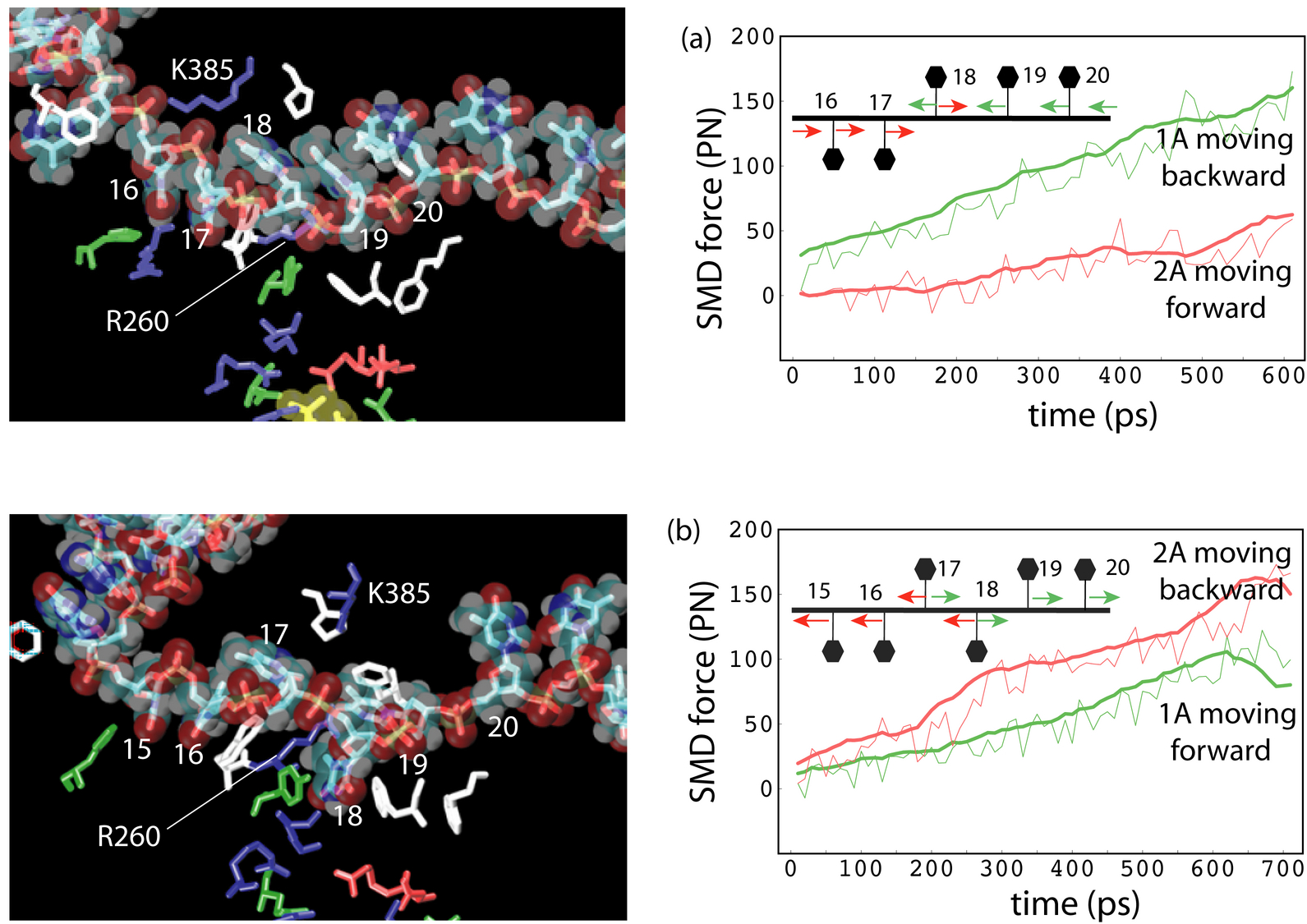
To find out the molecular mechenism of the ATP hydrolysis powered ssDNA translocation of PcrA, all-atom MD simulations were utilized, solving the classical Newtonian equations for a large number of atoms in a femotsecond time step. Using the program NAMD, MD simulations were performed on a fully solvated PrcA-DNA complex (see the middle configuration in the figure above) with and without ATP bound to the catalytic site. The system contained approximately 110,000 atoms and the simulations lasted for several nanoseconds. Since the translocation process actually happens on milliseconds time scale, the MD approach is limited. However, on the basis of these equilibrium simulations, interaction energies between the protein and the ssDNA nucleotides can be evaluated to estimate the effective potentials of the domain movements, which are to be utilized in the stochastic modeling of PcrA on a longer time scale. The simulations here provide us computational evidences at microscopic level an ``inchworm''-like model, that during the ATP hydrolysis cycle the domains 1A and 2A alternatively become weakly and strongly bound to ssDNA in such a manner that unidirectional translocation of PcrA along ssDNA results, see details in another recent publication .
 click for a larger view
click for a larger view Exemplary movies in the substrate (mpg, 2.1 MB) and the product (mpg, 2.1 MB) state
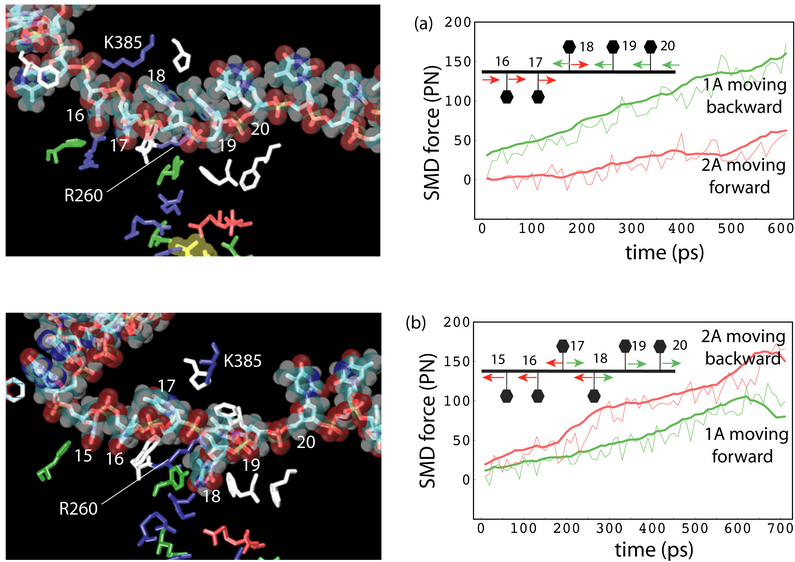
The time-scale gap presented between the equilibrium MD simulation and protein function level can be partially overcome by applying an external biasing potential, via, e.g.,steered molecular dynamics simulation (SMD). Although the approach still suffers from some limitatioins, such as requiring a priori insight into the most relevant motions, introducing artifacts due to insufficient relaxations, etc., it may reveal interesting dynamical properties in some cases. The SMD simulations, pulling the ssDNA segment through PcrA helicase in both forward and backward directions, also suggest that the domains 1A and 2A alternatively become weakly and strongly bound to ssDNA. The results are presented in the left figure.
Stochastic modeling on the unidirectional translocation
To reach the functional level description of PcrA translocation at milliseconds time scale, we need a very coarse grained modeling approach, and a simplest one is to replacing the two translocase domains 1A and 2A by two featureless beads. A straightforward way to characterize the movement of the beads along ssDNA is by means of the coupled Langevin equations describing the Brownian motions in a potential. Utilizing the effective potentials for the domain movments estimated from equilibrium MD simulations, the Langevin dynamics can be numerically solved and the corresponding non-equilibrium steady state behavior can be analyzed (see the publication ). Two limiting scenarios (the weak coupling and the strong coupling scenario) for the ATP hydrolysis coupled translocation are proposed in our modeling, depending on different coupling strength between the two domains 1A and 2A and different rate-limiting steps assumed; however, the physiologically relevant process may actually happen in a ``mixed'' scenario between the two. The stochastic modeling approach directly demonstrates that the unidirectional translocation is supported by the two domains alternatively experiencing higher and lower energy barriers along ssDNA.
Summary
Combining femtosecond to picosecond QM/MM calculation, nanosecond MD/SMD simulation and millisecond stochastic modeling, the ATP hydrolysis coupled ssDNA translocation of a monomeric PcrA helicase has been investigated. A rather comprehensive and detailed picture of PcrA translocation is revealed, e.g., a proton relay mechanism responsible for efficient ATP hydrolysis in the catalytic site and the means by which unidirectional motion along ssDNA is achieved. Similar strategy can be employed to investigate other molecular motors to reveal more about the mechanism by which these sophisticated nano-mechines perform their physiological functions.
Publications
Two posters at Biophysical Society Meeting 2006:
Investigators
Page created and maintained by Jin Yu.



{kind=link}
{kind=link}
{kind=link}