Molecular Dynamics of Viruses
Topics: Overview - STMV - Coarse graining - AFM nanoindentations of HBV - Atomic model of RHDV - Publications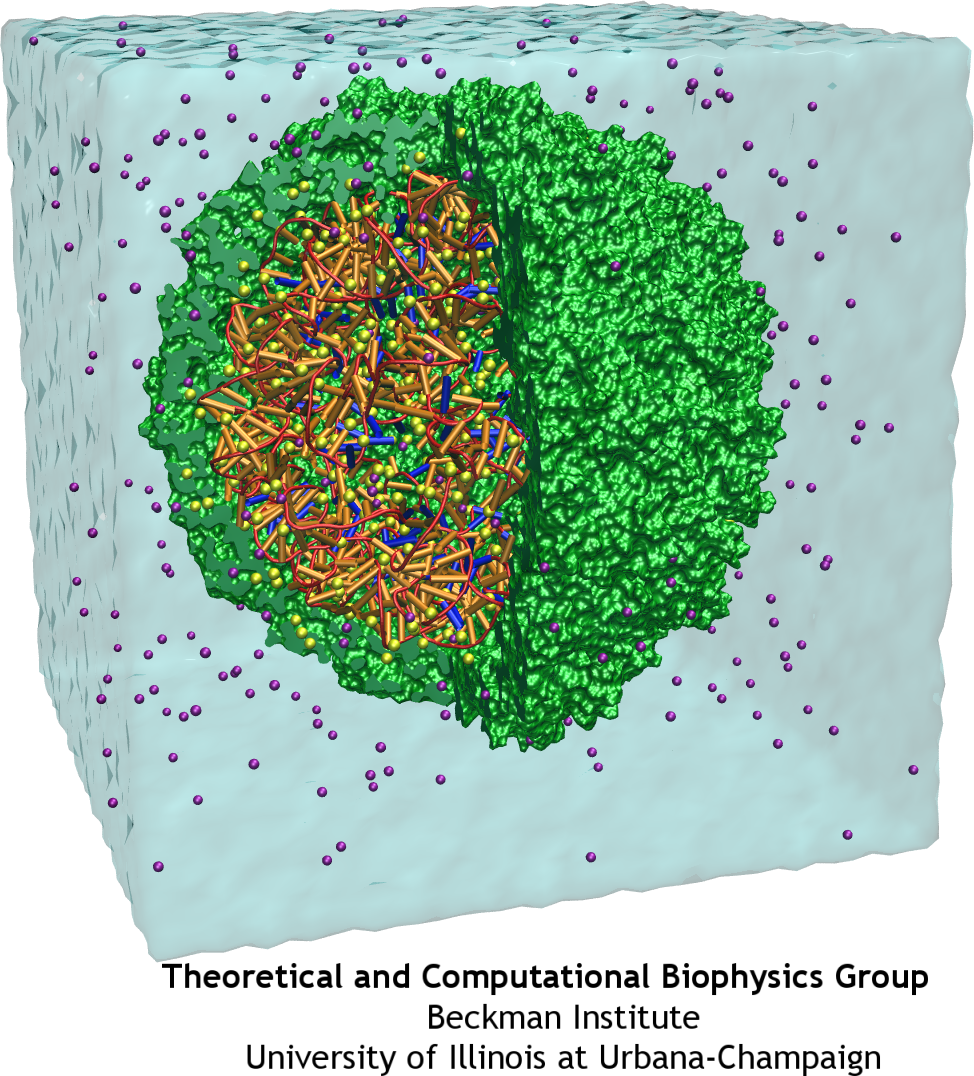
Viruses and Disease
Viruses are very small intracellular parasites that invade the cells of virtually all known organisms. They reproduce by utilizing the cell's machinery to replicate viral proteins and genomic material, generally damaging or killing the host cell in the process, and then the large number of newly generated viruses go on to infect other neighboring cells. Viruses are responsible for a wide variety of human diseases, ranging from the common (influenza and colds) to the exotic (AIDS and West Nile virus). Some viruses which are not dangerous to humans can also be exploited in technological applications, and viruses find use in genetic engineering applications and (increasingly) in the design of new nanomaterials.
At the very least, all viruses contain two components: the capsid (a protein shell), and a genome, consisting of either DNA or RNA. Some viruses also include accessory proteins to aid in infection, or a lipid bilayer to further protect their contents from the environment. The viral life cycle itself is fairly simple: viruses enter the cell, typically (but not always) through the interaction of their capsid with a receptor on the cell surface; the virus must then somehow disassemble its capsid to release its genetic material and any necessary helper proteins. The viral genome is then replicated and the proteins it codes for synthesized to produce the raw material for the production of many new viral particles; these new viruses then assemble, and then exit the cell either through the membrane or upon cell death.
Understanding the mechanics of the processes involved in the viral life cycle, and particularly the entry, disassembly, and new capsid assembly steps, at the molecular level will be the key to developing more effective treatments for viral diseases. Current antiviral drugs can target these steps (for example, by preventing the disassembly step from successfully occuring), but our knowledge of these steps is not yet sufficient for rational drug design. Thus, the Theoretical and Computational Biophysics Group studies the assembly and disassembly of several viruses using molecular dynamics, providing a time-resolved picture of the molecular processes involved in the viral life cycle.
Satellite Tobacco Mosaic Virus (STMV)
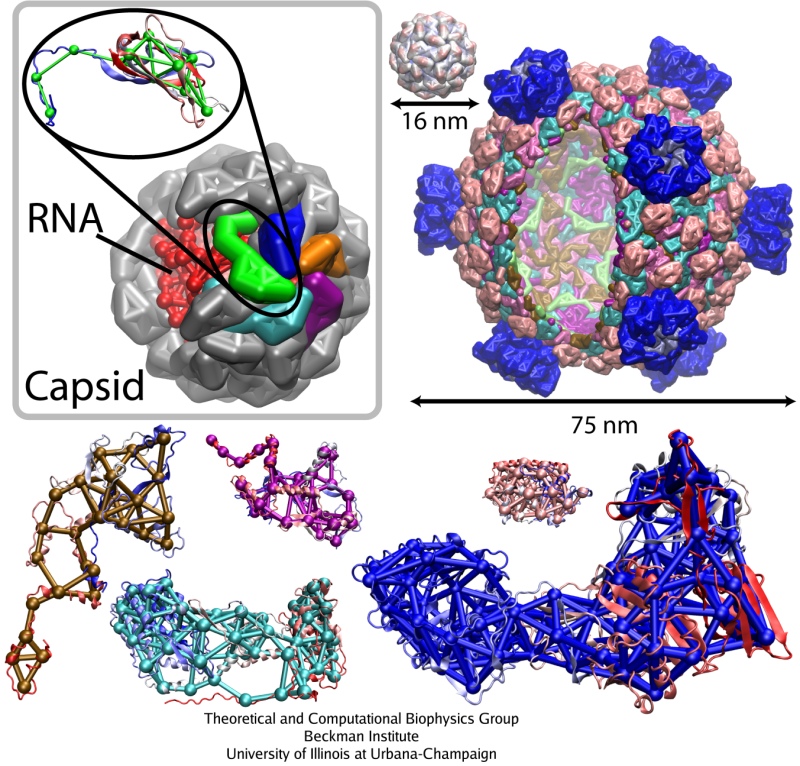
Satellite Tobacco Mosaic Virus is a small, icosahedral plant virus which worsens the symptoms of infection by Tobacco Mosaic Virus (TMV). Satellite viruses are some of the smallest possible reproducing units in nature; they achieve this by relying on both the host cell and a host virus (in this case, TMV) for the machinery necessary for them to reproduce. The entire STMV particle consists of 60 identical copies of a single protein that make up the viral capsid (coating), and a 1063 nucleotide single stranded RNA genome which codes for the capsid and one other protein of unknown function.
Mechanical Properties
The full crystal structure of the STMV capsid, along with a model backbone for the RNA it contains, has been solved by our collaborator Alexander McPherson (UC Irvine). It reveals that the capsid proteins of STMV form a perfect icosahedron, with three tightly associated monomers per face, and strong interactions between adjacent faces through interlocking tails. The RNA was found to form a set of 30 stem-loop regions, with each double stranded region aligning itself on the inner side of an edge of the capsid icosahedron. This highly ordered structure is held together through a combination of salt bridges between the positively charged protein and negatively charged RNA backbone, and a number of strong interactions between adjacent protein subunits. Because of the interlocking nature of the capsid subunits and the strength of the fully formed structure, the STMV capsid is sometimes referred to as "chainmail."
Questions about STMV
Although the STMV crystal structure has revealed a great deal of information about STMV, some unresolved questions remain regarding this virus. The mechanism through which the virus assembles has not yet been discovered through experimental techniques; among the most critical questions is whether STMV assembly is primarily driven by the capsid or the viral RNA. In addition, it is quite unclear how the virus ever disassembles once it has been taken into a host cell, because of the very high stability of the capsid's structure. Although a crystal structure for the RNA backbone exists, it is also unclear how the sequence of the virus maps onto that backbone, because it has been impossible to obtain a high enough resolution electron map for that part of the structure. These questions regarding STMV are critical not only for understanding this particular virus, but for understanding the mechanisms of viral assembly and infection in general; STMV merely provides a simple, convenient system for exploring these properties of viruses.
STMV makes an especially good candidate for study using molecular dynamics (MD) because it is relatively small for a virus; the fully solvated STMV system shown above consists of approximately 1 million atoms, which is on the high end of what is feasible to simulate using traditional molecular dynamics today. Molecular dynamics simulations provide both a visual representation and quantitative data regarding the thermal motion of a system's structure over time, or its response to applied forces. Although the full processes of assembly and disassembly are too slow to be fully simulated, brief molecular dynamics simulations on the STMV structure under various conditions may yield important insights into the processes under study.
Stability of STMV components
Molecular dynamics simulations on the complete STMV structure have shown that it is perfectly stable on a time scale of tens of nanoseconds (mpg movie, 22M); in the process, it was discovered that because the capsid is not perfectly rigid, the most prominent modes of motion in the subunits arise in waves along an equatorial region, rather than the radial oscillations that one might expect given its structure (publication 1, below). Both to test the physical properties of the viral components and to investigate possible mechanisms for assembly, the capsid and RNA were also simulated independently of one another.
When simulated alone in solution, the RNA genome of STMV proved to be remarkably stable; it expanded in width by roughly 15% but showed no major changes in secondary structure or the relative positions of secondary structure elements. The STMV capsid alone, in contrast, proved to be quite unstable, and showed a remarkable collapse over the first 5-10 nanoseconds of simulation (mpg movie, 15M). These results were verified through three separate MD runs; in each simulation the capsid collapsed, albeit at a different place. The reason for the collapse in the absence of RNA seems to be that there are a number of potential interactions between dimers of the STMV capsid protein that are stronger than the usual interactions in the fully formed capsid, but which in the complete structure are blocked by the presence of the RNA. This result is especially interesting when considered in the context of virus assembly, as it suggests that it must be the RNA, and not the protein, which nucleates assembly into the complete icosahedral particle; it is possible that the RNA by itself forms a somewhat spherical shape, and then protein monomers or dimers attach to it and pull things into the final icosahedral configuration.
Future Work
In an effort to address the questions above, we are continuing MD studies on the STMV particle to obtain more evidence on how the particle may assemble. In addition, simulations under variable pH conditions will be used to observe how the protonation state of residues in the protein affects the stability of the structure; this is particularly relevant to viral disassembly, as it may represent a mechanism for destabilizing the capsid enough to release the RNA.
Dynamics of Viral Capsids Described by Coarse-Grained Modeling

One of the most important roles of a viral capsid is to protect its contents, but the capsid also needs to become unstable to release the virulence factors into the host cell. Such transitions are determined by the dynamics of protein units that comprise the capsid, but the spatial and temporal resolution of experiments is usually much lower than required to distinguish the movements of individual capsid proteins. On the other hand, all-atom simulations of a whole virus are difficult too, due to the huge size of the system. One solution that allows to discern movements of individual proteins, and still reach desired length and time scales, is to reduce the number of degrees of freedom considered in numerical simulation, an approach known as ``coarse-graining'' (CG). A method called ``shape-based coarse-graining'', developed by TCBG scientists, was applied to study a number of viral capsids, revealing a variety of behaviors depending on the strength of interactions between the capsid proteins.
In the shape-based coarse graining method, the shape of a protein is reproduced by several point masses, which interact through effective potentials. For the study of viruses, the ratio of about 200 atoms per CG particle was used. This level of coarse-graining allows one to reach the microsecond time scale. Notably, the employed CG method has been developed in a general form, such that it can be applied readily to systems other than viral capsids. This same method was applied to study the dynamics of the bacterial flagellum, using the ratio of 500 atoms per CG particle.
Testing the coarse-grained model: comparison with the all-atom study
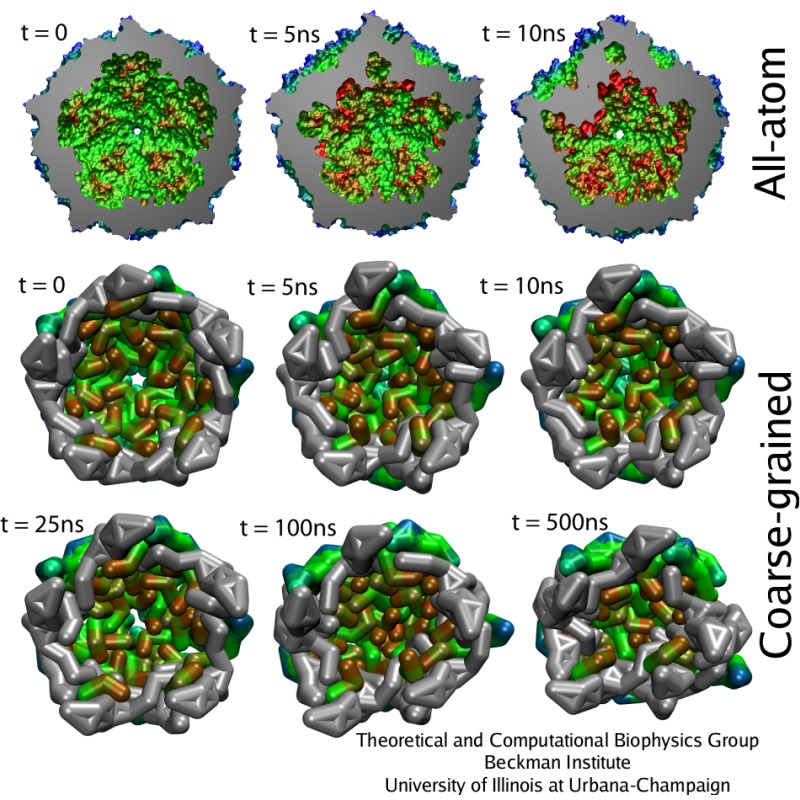
The previous all-atom study of STMV represented the best case for the new CG model to be tested. It was found that the CG simulations reproduced the results of the all-atom simulations quite well. For example, the radial distribution of mass, the average and minimal distances between the center of the virus and capsid proteins monomers, and the radial rmsd were within few Angstroms from those values in the all-atom study. This should be compared to the thickness of the capsid, which is about 35 Angstroms, and to the fact that each CG bead represents a part of the protein 10-20 Angstroms in size. Furthermore, the CG simulations also reproduced the patterns and timescales of the collapse observed for the STMV capsid without the RNA. Thus, not only the shape of STMV is described well by the CG model, but also the dynamics and characteristic times of the dynamics are very similar between the all-atom and CG levels of description, suggesting that the timescales of motions observed in CG simulations of other viruses are realistic.
Simulating the dynamics for a variety of capsids
What determines how rigid the capsid is? To answer this question, a icosahedral capsids of a number of viruses were simulated, with the sizes from 17 to 75 nm, over times of 5 to 25 microseconds. Presently, all-atom simulation of any of these viruses on a microsecond timescale would be technically impossible; the CG technique is therefore playing here a role of the bridge that helps to transfer the knowledge about the atomistic structure of the studied system into the knowledge about the large-scale movements of the system.
Among the viruses chosen for the study were three satellite plant viruses: STMV, the satellite panicum mosaic virus (SPMV), and the satellite tobacco necrosis virus (STNV). About 20 nm in diameter, each of these viruses consists of a short (~1000 nucleotides) RNA genome packed inside a T=1 capsid composed of 60 identical proteins. They infect plants, relying on the pre-infection of a plant by their respective helper viruses. Next choice was the brome mosaic virus (BMV), a T=3 plant RNA virus about 28 nm in diameter, composed of 180 identical protein units. Then, three larger and more complex assemblies were simulated, the poliovirus capsid, the bacteriophage phiX174 procapsid, and the reovirus core. Poliovirus and reovirus infect humans, whereas bacteriophage phiX174 infects bacteria. The poliovirus capsid, 33 nm in diameter, is composed of 60 copies of four different polypeptides. The bacteriophage phiX174 procapsid (T=1 icosahedron, 36 nm in diameter) is a precursor particle formed during the assembly of the virus; the viral DNA is actively injected into the procapsid. This assembly is built up of four proteins, with 60 copies of three of them and 240 copies of the fourth one (in four distinct conformations). Finally, the reovirus core is one of the largest viral structures available, about 75 nm in diameter; it is a T=1 structure composed of five different proteins. Altogether, 480 protein units comprise the particle. This assembly represents a biochemical machine performing the synthesis, modification and export of the viral messenger RNA.
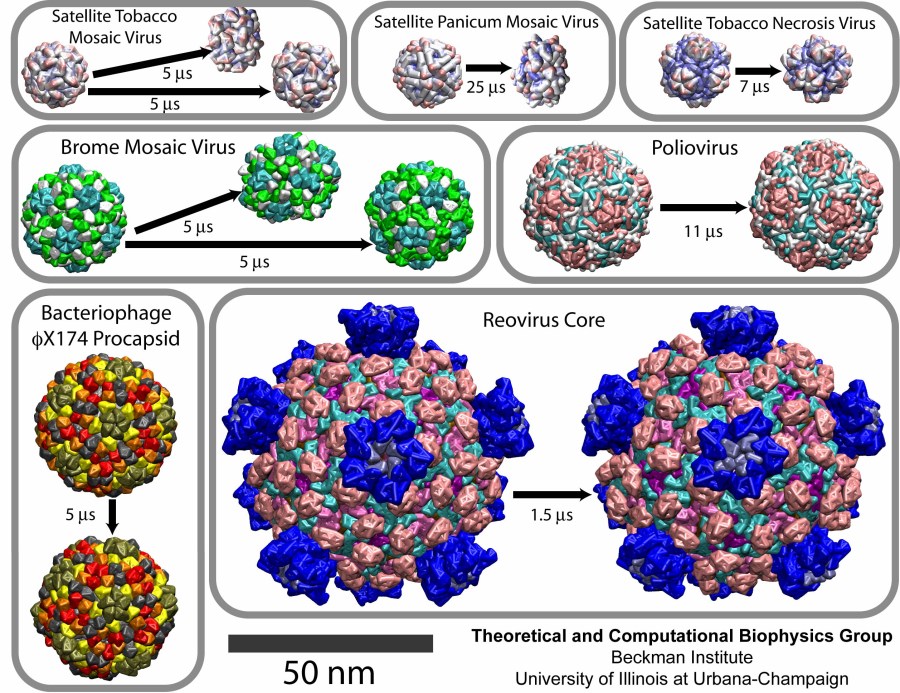
The CG simulations revealed a range of flexibilities for the studied capsids. The empty capsids of the three satellite viruses, STMV, SPMV, and STNV, collapsed, as was the case for the all-atom study of STMV. The complete STMV virion, however, was stable, again in agreement with the all-atom simulation.
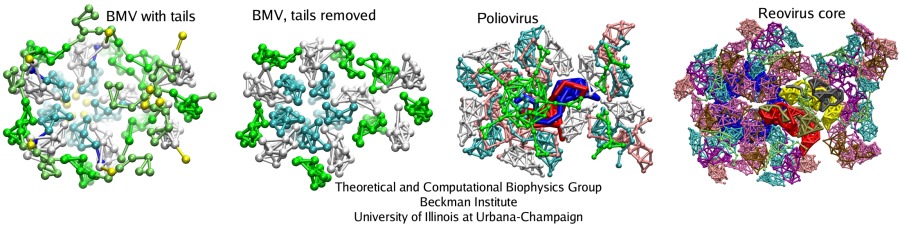
The capsid of BMV was stable overall, but very flexible, and indeed, empty BMV capsids have been observed experimentally. On the other hand, numerous experiments illustrated that a cleavage of the N-terminal tails of the BMV capsid proteins results in the capsid falling apart or restructuring into various possible conformations. A CG model of the BMV capsid with the N-terminal tails removed was built, and in the resulting simulation the capsid collapsed. The initially spherical capsid was brought into a discoidal shape.
CG simulations of the reovirus core, the bacteriophage phiX174 procapsid, and the poliovirus capsid demonstrated that these are stable structures. The quantitative analysis showed that the poliovirus capsid is the most flexible among these three systems, the next one being the phiX174 procapsid, and the most rigid one being the reovirus core.
Movies for each of the simulations can be downloaded here: STMV, 9.1M; SPMV, 5.4M; STNV, 6.9M; BMV, 8.4M; poliovirus, 9.9M; bacteriophage phiX174, 7.2M; reovirus core 4.8M.
Interlocking of unit proteins determines the flexibility and stability of a capsid
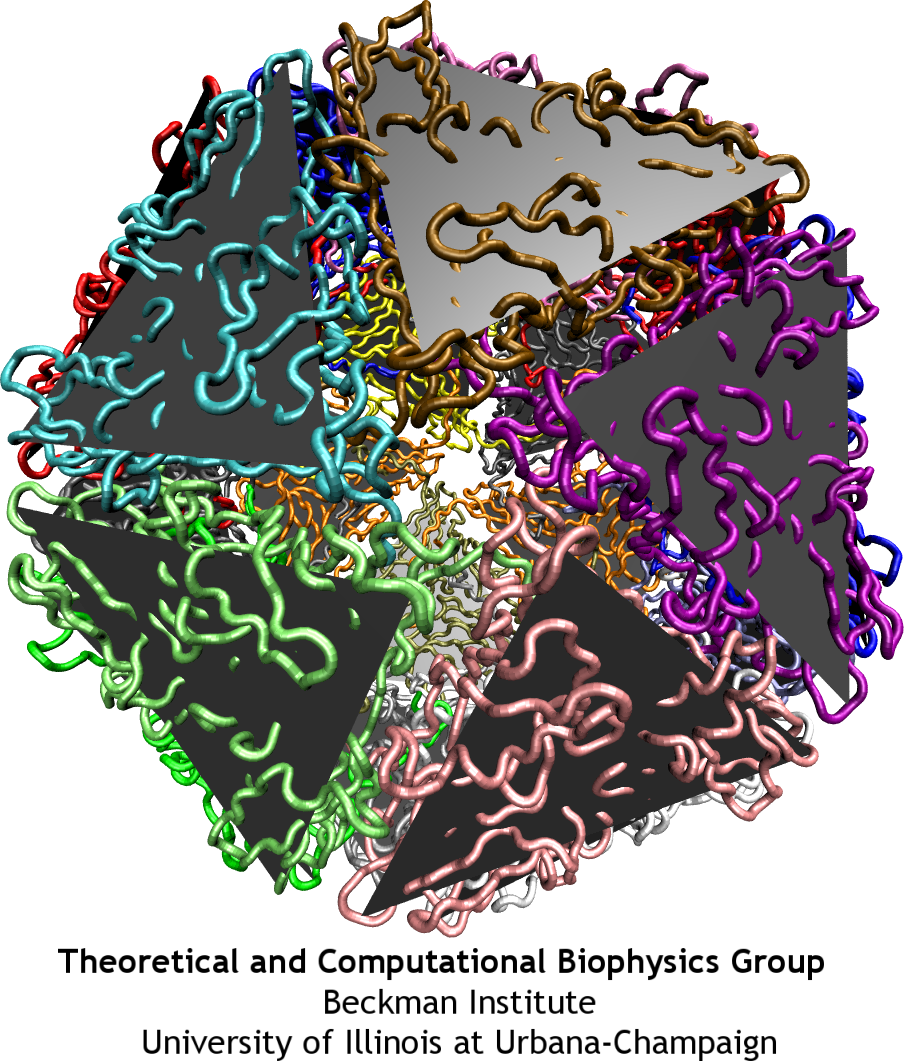
Analysis of interactions between the protein subunits in the capsids of the studied viruses shows that the stability and flexibility of the capsid is closely correlated with the strength of these interactions. Larger capsids, such as the reovirus core, have more proteins, which intricately intertwine with each other, featuring even a ``thread and needle'' arrangement. In the smaller capsids, such as the one of STMV, unit proteins generally only touch each other by the edges. Examples are shown in the figure below: parts of the capsids are depicted there as seen from inside. Stability of the BMV capsid, as our study have shown in agreement with the experiments, is determined by the entanglement of the N-terminal tails of the capsid proteins. When the tails are removed, almost half of the attractive energy is lost, and the capsid becomes unstable. The poliovirus capsid and reovirus core have more protein units, that are arranged in a few intertwining layers, making these structures very rigid.
With more contacts between the protein units, a capsid has more hydrogen bonds and salt bridges per area unit, and the frictional force between the capsid faces rises; these factors enhance the stability. The all-atom and CG simulations suggest that the viruses like STMV, SPMV, and STNV have too few contacts between the capsid subunits, so that their capsids are unstable without the genetic material. The genetic material (RNA) plays a foremost structural role for these small viruses, stabilizing the protein units, which interact strongly with the RNA by their long tails that reach deep towards the center of virions. Larger structures, such as BMV and poliovirus capsids, bacteriophage phiX174 procaspid, and reovirus core, establish their stability through a hierarchy of contacts between the protein units themselves, the size of these assemblies allowing for many contacts to be created.
The way a virus maintains its stability determines the assembly and disassembly pathways; as our study shows, the all-atom and CG simulations of macromolecular complexes are now becoming capable of gradually elucidating the properties underlying these amazing phenomena.
AFM nanoindentations of the hepatitis B virus capsid
The mechanics of viral capsids and their protein components plays a key role in the replication cycle of viruses, since a capsid should be stable to protect the virus from a hostile environment outside of host cells, but also needs to disassemble or otherwise release the viral genome at the onset of the infection process. Recently, mechanical properties of several viruses have been elucidated through AFM nanoindentation experiments, where the force response of a capsid to an AFM indentation is recorded in the form of a force-indentation (FZ) curve. However, molecular details underlying the observed capsid mechanics remain mostly unresolved. This problem can be overcome through MD simulations, but at present, for most viruses a CG approach would be required, due to the large size of their capsids. The shape-based coarse-graining has been applied together with AFM nanoindentation experiments to study the mechanics of the empty capsid of the human hepatitis B virus (HBV), as reported by Arkhipov et al. (2009). The HBV capsid is studied in its T=4 form, for which a crystal structure is available (average radius R=14.5 nm). Previous AFM experiments on HBV showed that for low forces the indentation is reversible. However, for higher indentations irreversible deformations occur, and with CG simulations we have been able to clarify the molecular mechanism behind this irreversibility.
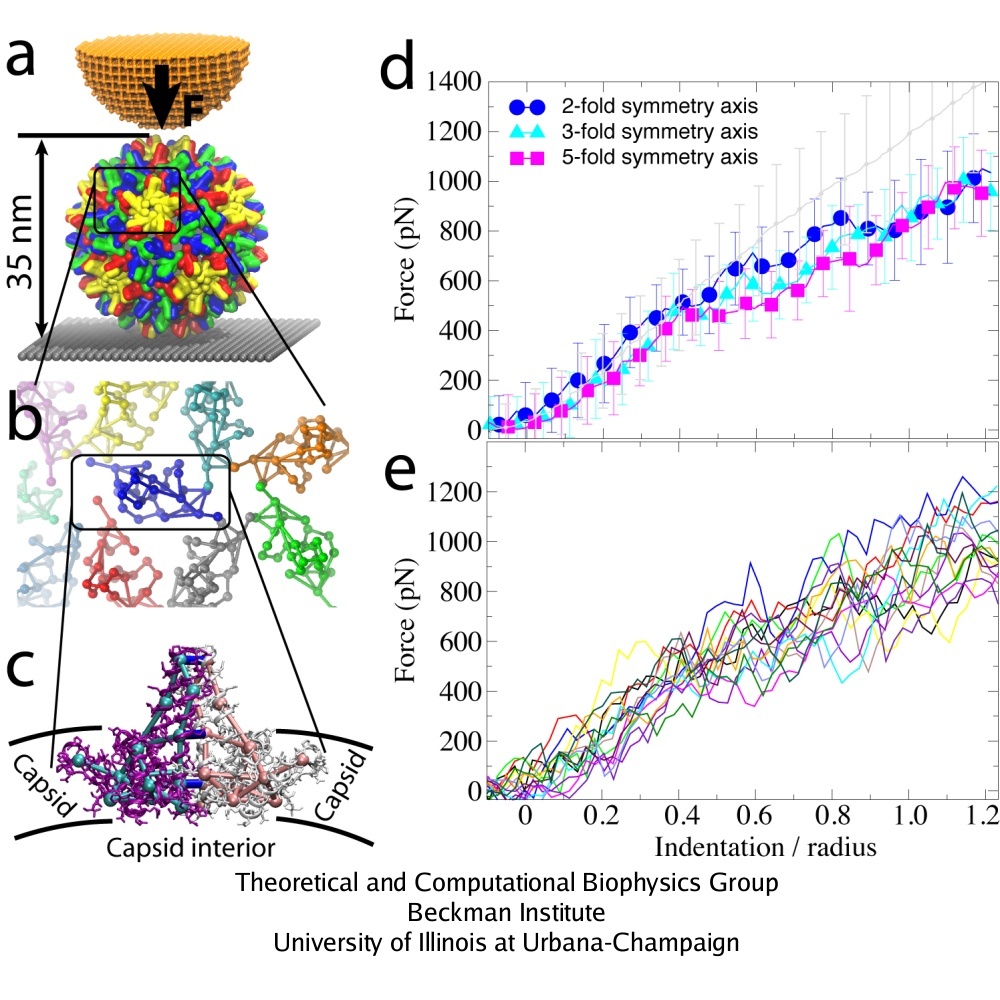 AFM nanoindentations of the HBV T=4 capsid. (a) The capsid is attached to a surface and squeezed by the AFM tip. (b) Arrangement of protein dimers on the capsid surface. (c) Within a dimer, the monomers (purple, white) are connected via extensive surface contacts. All-atom and CG models are shown. (d) Force-indentation curves averaged over all simulations for each pushing direction (color), and over all experiments (gray). (e) Force-indentation curves for individual simulations, each shown by a different color.
AFM nanoindentations of the HBV T=4 capsid. (a) The capsid is attached to a surface and squeezed by the AFM tip. (b) Arrangement of protein dimers on the capsid surface. (c) Within a dimer, the monomers (purple, white) are connected via extensive surface contacts. All-atom and CG models are shown. (d) Force-indentation curves averaged over all simulations for each pushing direction (color), and over all experiments (gray). (e) Force-indentation curves for individual simulations, each shown by a different color.
SBCG simulations were performed for maximal indentation depths Z = 0.35 R (5 nm) and Z = 1.25 R (18 nm, which is the outer capsid radius). For both maximal indentation depths, five simulations were carried out with pushing along the 5-fold symmetry axis of the capsid, five along the 3-fold, and five along the 2-fold axis. Three repeated rounds of pushing were done in each simulation. The indentation of 1.25 R was reached within approximately 10 microseconds. Dynamics of the capsid under AFM nanoindentation is shown in the movies: mpg movie of a simulation with Z = 0.35 R, 3.1M, and mpg movie of a simulation with Z = 1.25 R, 3.8M

A remarkable agreement between simulation and experiment was found, particularly for indentations of up to 0.4 R. For simulations with three rounds of pushing performed, one observes reversible behavior of the force response for Z = 0.35 R, while for Z = 1.25 R the capsid deformation is clearly irreversible. This result agrees with experiments, which show that the HBV capsid deforms reversibly if the indentation is stopped at Z = 0.35 R to 0.55 R, and irreversibly for larger indentations. In both simulation and experiment for Z = 1.25 R, the force response reduces from one round to the next, so that at the maximal indentation it is 100-200 pN smaller than in the first round. However, while the simulated curves show that the capsid height remains reduced after the AFM tip retracts, experimental curves indicate nearly full capsid height recovery in each round. Most likely, this is due to the difference in the relaxation time between the pushing cycles, which is about 1 s in experiments and only a few microseconds in simulations. However, on closer inspection a capsid deformation persisting after tip retraction is evident in the experiment as well, where one finds that after 5 pushing cycles the capsid height decreases by approximately 10 %.
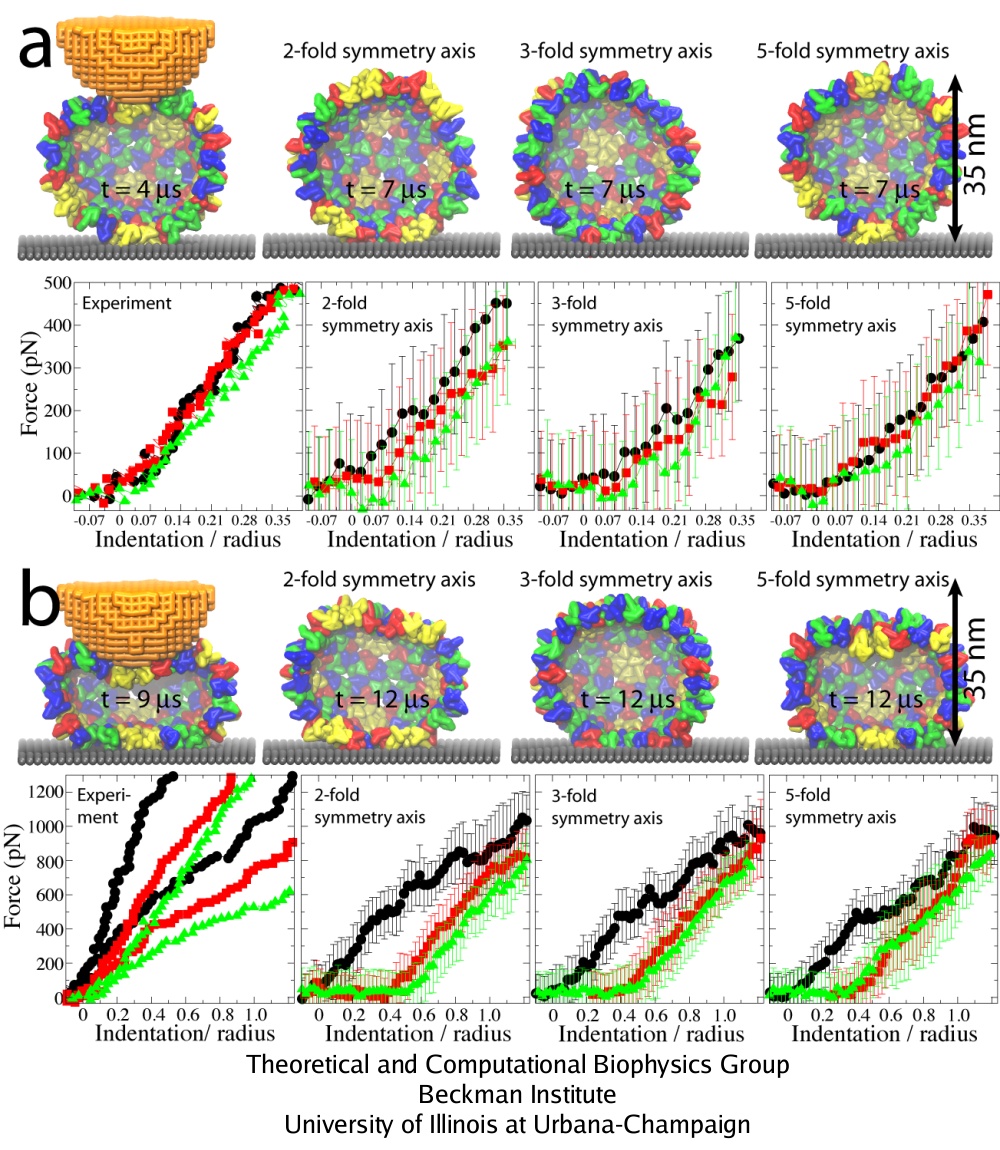 Three rounds of repeated pushing. (a) Pushing with Z = 0.35 R. (b) Pushing with Z = 1.25 R. The images of the capsid on the left show conformations at the time of the maximal indentation; the remaining images show the capsid during relaxation. FZ curves: 1st round, black; 2nd round, red; 3rd round, green. The experimental curves show indentations of individual capsids. In the left plot in (b), results of two independent experiments are shown.
The curves labeled as ``2-'', ``3-'', and ``5-fold symmetry axis'' are the simulated curves averaged over all simulations for each pushing direction.
Three rounds of repeated pushing. (a) Pushing with Z = 0.35 R. (b) Pushing with Z = 1.25 R. The images of the capsid on the left show conformations at the time of the maximal indentation; the remaining images show the capsid during relaxation. FZ curves: 1st round, black; 2nd round, red; 3rd round, green. The experimental curves show indentations of individual capsids. In the left plot in (b), results of two independent experiments are shown.
The curves labeled as ``2-'', ``3-'', and ``5-fold symmetry axis'' are the simulated curves averaged over all simulations for each pushing direction.
What is the origin of the observed irreversibility of capsid deformation? Analysis of how interactions between protein units of the capsid change throughout simulations shows that bending and shifting of these units, along with establishment of contacts between capsid and substrate, are the main contributing factors. One finds, however, that the arrangement of protein units is essentially unaffected by the indentation, i.e., the local structure of the capsid remains the same, even though the capsid is strongly deformed globally. The position and orientation of units with respect to each other are not significantly affected, but their local bending and slight rearrangement is enough to introduce new favorable interactions that lock the capsid in the deformed state, once certain depth of indentation is passed. Thus, the irreversibility is due to capsid-substrate interactions, capsid protein bending, and slight shifting of these proteins with respect to each other, rather than from global rearrangement.
The HBV T=4 capsid is a relatively small, spherical, and homogeneous virus shell, with loose protein packing. Capsids of many other viruses exhibit a capsomer organization with extensive protein-protein contacts, which greatly affect the mechanical stability of the capsids. The study of HBV suggests that these differences in capsid protein organization underlie the differences stiffness of various viral capsids, as recorded in AFM experiments. Namely, experiments show that the HBV capsid, as well as the similarly loosely packed CCMV capsid, are less stiff than the capsids of such viruses as MVM and bacteriophages lambda and phi29. Furthermore, the lambda and phi29 capsids break at indentations of 20-25 % of their radius and show material fatigue under repeated small indentations. In contrast, irreversible deformations arise at 60 % of R for HBV, and HBV capsids do not show signs of fatigue after tens of shallow indentations. One finds that large-scale deformations of the HBV capsid occur without disruptions of local capsomer order, i.e., without capsid ``failure'', and that slight changes in this order and bending of capsomers are enough to maintain the capsid deformation after the load is removed. For viruses such as MVM and bacteriophages lambda and phi29, the deformation scenario should be different, as the capsomers are interlocked with each other, leading to a stiffer capsid and more local deformations upon force application than in the case of HBV. The tight interlocking is also difficult to restore once it is broken, resulting in a stiff, but relatively brittle capsid and in capsid ``failure'' upon strong deformation; the weak surface contacts of HBV capsomers make the HBV capsid rather more plastic and allow the capsid geometry to undergo smooth transitions.
Atomic Model of Rabbit Hemorrhagic Disease Virus (RHDV)
Rabbit hemorrhagic disease (RHD) is extremely contagious in adult rabbits and is often associated with liver necrosis, hemorrhaging, and high mortality. It was first described in China in 1984, and within a few years had spread worldwide. RHD outbreaks still occur on almost every continent and cause significant mortality rates, being endemic in Europe, Asia, Africa, and Australia. This disease has a significant impact on the rabbit industry and ecology. The etiological agent of RHD is rabbit hemorrhagic disease virus (RHDV), which has a single-stranded, positive-sense, polyadenylated RNA genome of ~7.5 kb. Mature RHDV virions are spherical, non-enveloped particles with a T = 3, icosahedral capsid whose outer diameter varies between 32 and 44 nm and whose structure is defined by characteristic, cup-shaped depressions. The only capsid protein present in RHDV, VP60, is composed of three domains, which include the N-terminal arm (NTA), the shell (S), and the protrusion (P), the latter of which is further divided into P1 and P2 sub-domains.
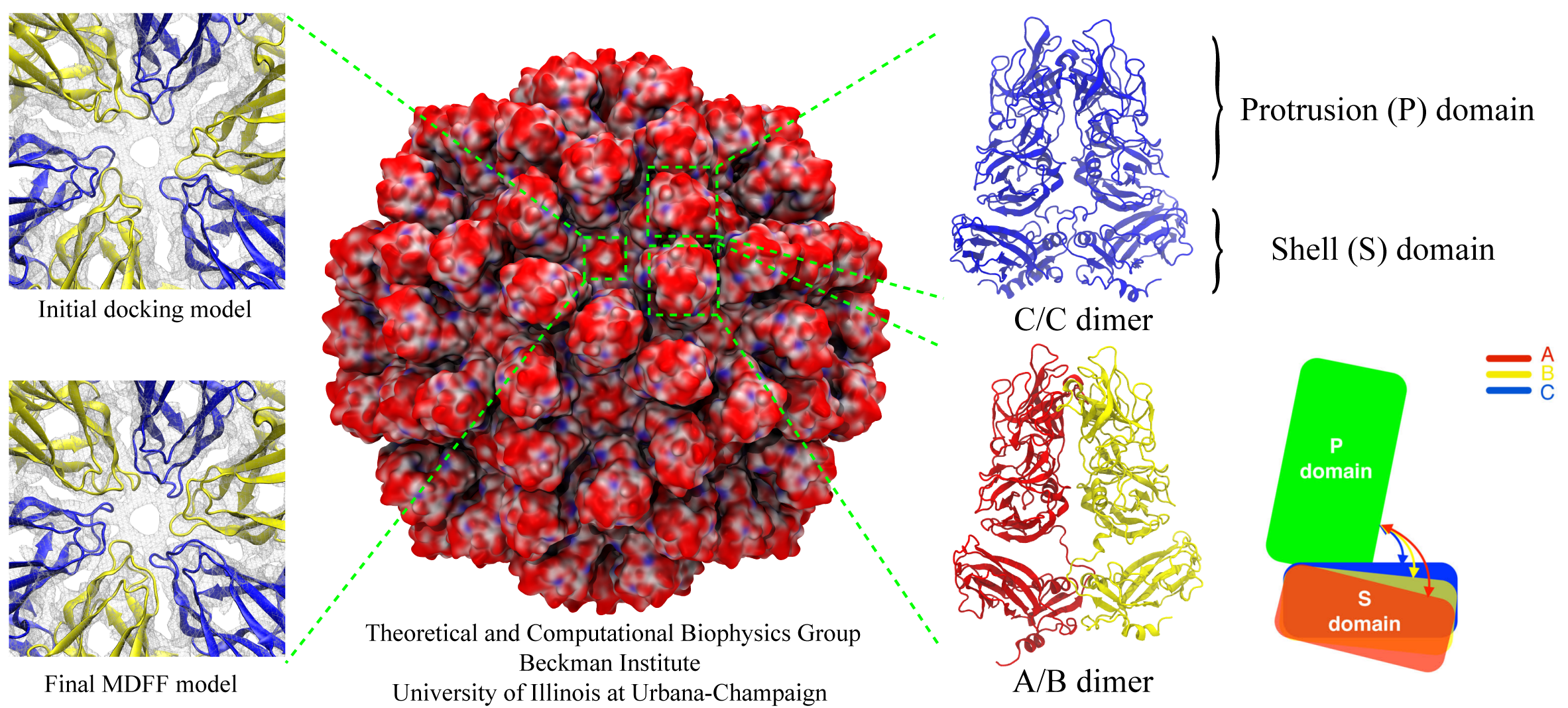
The asymmetric unit of the RHDV capsid consists of three quasi-equivalent VP60 subunits (A, B and C). The 180 VP60 subunits that comprise the capsid are organized as 90 dimers, each of which appears as an arch-like capsomer. Thirty C/C capsomers are located at the icosahedral two-fold symmetry axes and the remaining 60 A/B capsomers are located at pseudo (local) two-fold axes. Three A/B and three C/C dimers are positioned in alternate fashion around each icosahedral three-fold axis to form pseudo-six-fold arrangements, and five A/B dimers encircle each five-fold axis. Together, these capsomers produce a contiguous shell and 32 cup-shaped, surface depressions, the latter of which are a characteristic feature of the structure of all caliciviruses.
Our collaborator Fei Sun (Institute of Biophysics, Chinese Academy of Sciences) has obtained the crystal structure of the shell (S) and the protrusion (P) domains of RHDV major capsid protein, VP60, each at 2.0 Å resolution, along with a cryo-electron microscopic (EM) reconstruction of wild-type RHDV at 6.5 Å resolution. An ab initio model of the N-terminal arm (NTA) was built based on the high-resolution (5.5 Å) cryo-EM density of RHDV inner shell. Integrating all these structural data, an atomic model of RHDV was built through Molecular Dynamics Flexible Fitting (MDFF) method developed here at Theoretical and Computational Biophysics Group (TCBG). The MDFF procedure improved the overall fitting of the crystal structure to the cryo-EM density. Notably, MDFF closed the gaps between B and C subunits at the interface around the 3-fold axis. Structural comparisons among the A, B and C monomers of the MDFF-refined model, when aligned to the P domains, revealed that large conformational changes accompany relative movements and rotations of the S domain with respect to the P domain. Sequence alignments of VP60 from six groups of RHDV isolates revealed seven regions of high variation that could be mapped onto the surface of the P2 sub-domain and suggested three putative pockets might be responsible for binding to histo-blood group antigens. A flexible loop in one of these regions was shown to interact with rabbit tissue cells. Subsequent animal-based experiments confirmed that synthetic peptides derived from this loop stimulate production of high-titer antibody that can protect rabbits from RHDV infection. Our pseudo-atomic model of RHDV provides a structural framework for developing new anti-RHDV vaccines and will also help guide use of the RHDV capsid as a vehicle to display human tumor antigens as part of anti-tumor therapy.
PublicationsInvestigators
Collaborators
Page created and maintained by Anton Arkhipov and Peter Freddolino.


