DNA methylation and hydroxymethylation
DNA methylation
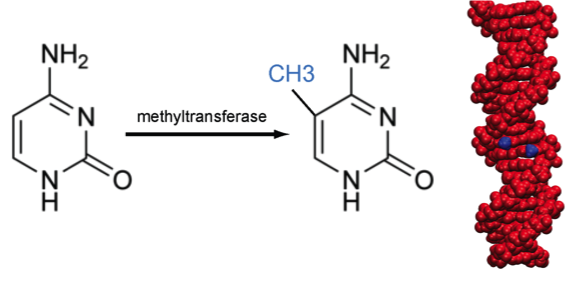
DNA methylation
Methylation of cytosine is a covalent modification of DNA, in which hydrogen H5 of cytosine is replaced by a methyl group. In mammals, 60% - 90% of all CpGs are methylated. Methylation adds information not encoded in the DNA sequence, but it does not interfere with the Watson-Crick pairing of DNA - the methyl group is positioned in the major groove of the DNA. The pattern of methylation controls protein binding to target sites on DNA, affecting changes in gene expression and in chromatin organization, often silencing genes, which physiologically orchestrates processes like differentiation, and pathologically leads to cancer [reference see publications below].
Biological functions of DNA methylation
- Transcriptional gene silencing
- Genome stability
- Chromatin compaction
- Genome defense
- Suppression of homologous recombination between repeats
- X chromosome inactivation (females)
- (Nature Genetics 38:1359 (2006))
 Same Genes but a Different Kink in the Tail. PLoS Biol 1:3 (2003) (Photograph courtesy of Emma Whitelaw, University of Sydney, Australia.) |
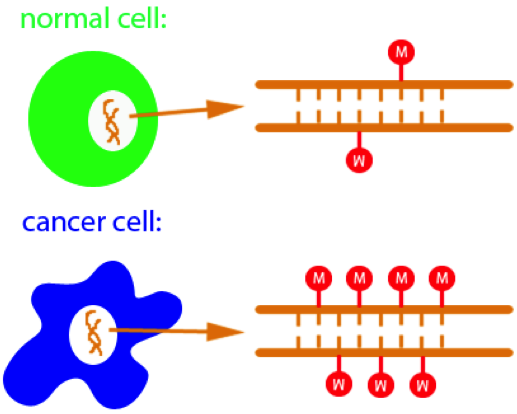 Hypermethylation of CpGs leads to cancer |
DNA hydroxymethylation
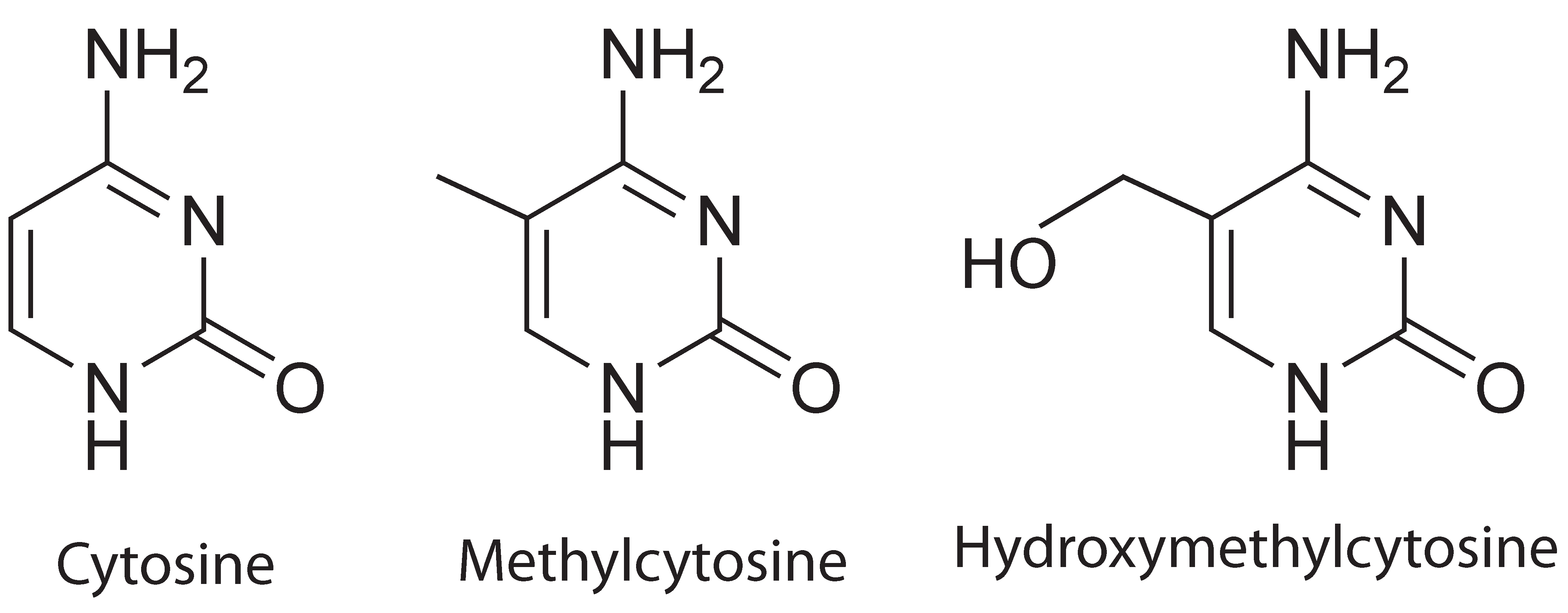
Comparison of cytosine, methylcytosine and hydroxymethylcytosine
Cytosine hydroxymethylation was recently discovered as another important epigenetic modification on DNA in mammalian cells. Similar to methylation, hydroxymethylation replaces, at the C5-position in cytosine, the hydrogen atom by a hydroxymethyl group. It has been demonstrated that cytosine hydroxymethylation is also involved in gene regulation. For example, the hydroxymethylation level has been found to be associated with pluripotency of stem cells. Disturbed hydroxymethylation of DNA cytosine can result in disordered cell functions, causing different types of cancers, e.g., myeloid cancers [reference see publications below].
Using nanopore to detect DNA methylation
We have reported measurements and simulations of the permeation of methylated DNA through a synthetic nanopore, using an electric field to force single molecules to translocate across the membrane through the pore (see also our nanopore site). We observe a voltage threshold for permeation of methylated DNA through a < 2nm diameter pore, which we attribute to the stretching transition of DNA, occurring in such pore induced by the voltage gradient; the degree of stretching can differ by >1V/20nm depending on the methylation level, but does not depend sensitively on DNA sequence. The threshold for squeezing unmethylated DNA through 1.8 nm pore is about 3.6 V while fully-methylated DNA shows a threshold 2.7 V. In MD simulation, we recognized a significant difference between the translocation speeds of methylated and unmethylated DNA, namely, speeds of 1.0 nm/ns and 0.8 nm/ns, respectively. The velocity difference implies that methylated DNA passes the pore more readily than unmethylated DNA, which indeed is consistent with the threshold voltage difference measured. Conformations of the DNAs are shown in the figure below. One can recognize that both translocating DNAs exhibit a B-form structure. However, methylated DNA is considerably more ordered and adheres more closely to the ideal B-form than does unmethylated DNA. More detail can be found here.
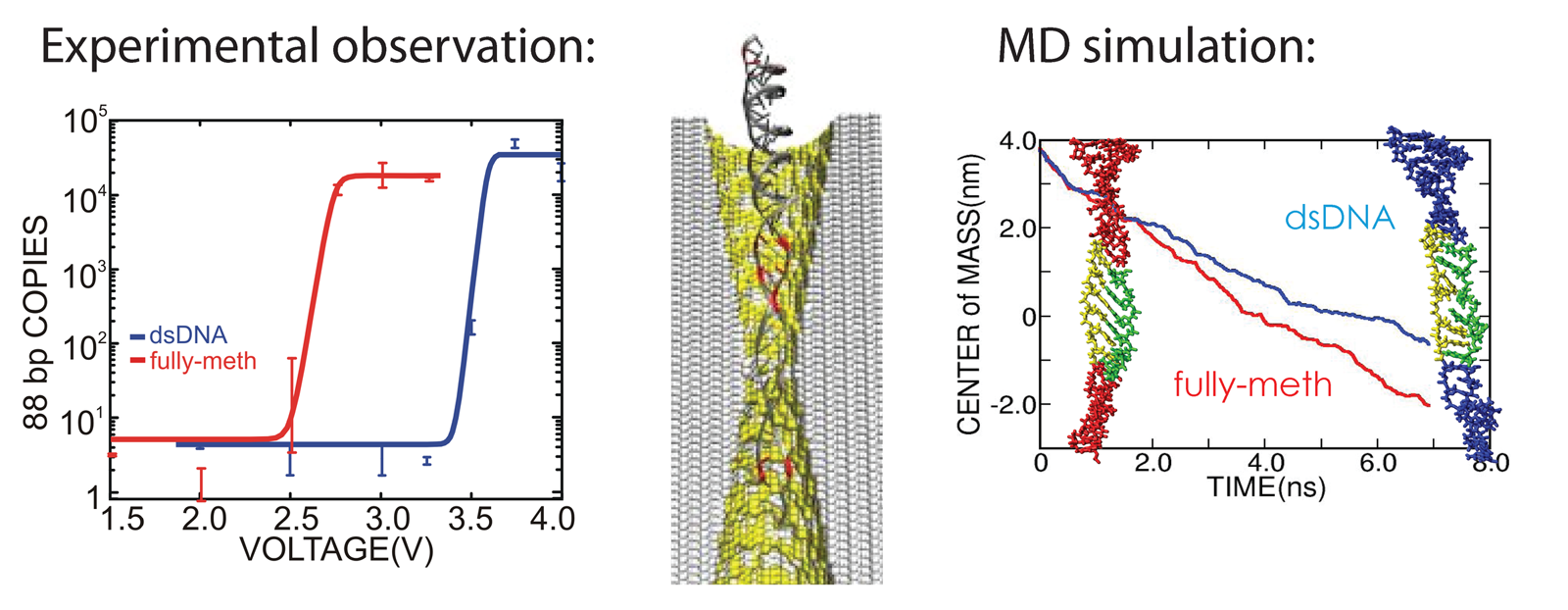
Using nanopore to detect DNA methylation
DNA methylation affects strand separation
The role of cytosine methylation in epigenetics is generally understood to involve effects of DNA methylation on chromatin structure or on the action of transcription factors, either one effect controlling in a long-time manner gene expression levels and, thereby, cell differentiation. The small effect of cytosine methylation on DNA thermodynamic properties let one to believe that a change of DNA physical properties by themselves, i.e., not ones mediated by the nucleosome and transcription factors, do not need to be considered for a role in epigenetics. Our study shows now that a role of methylated DNA physical properties needs to be reconsidered as a direct epigenetic control factor. We found through novel and extremely extensive force measurements comparing non-methylated and various methylated DNAs that methylation affects the propensity for mechanical DNA strand separation to a significant degree.
Determination of the mechanical stability of methylated DNA by molecular force assay measurements. Measurements stretching DNA mechanically to the point of strand separation were carried out with a new chip technology, molecular force assay (MFA), that permits one to stretch and rupture apart thousands of DNA double strands organized by methylation type on spots on the chip with a fluorescent signal readout. The experimental setup of the MFA at a molecular level consists of molecular force probes (MFPs). The MFPs are composed of two DNA duplexes, a target duplex 1 • 2 and a reference duplex 2 • 3, which are coupled in series and connected between two surfaces. Here, the target DNA duplex is 20 bp long and contains zero (nDNA), one (mC-1c-DNA) or three (mC-3-DNA) 5-methylcytosines (mC) per strand, while the reference duplex is the same for all three different MFPs. About 104 MFPs per μm2 are anchored in parallel between a glass slide (lower surface) and a PDMS stamp (upper surface). The different MFPs are immobilized as well separated spots on the glass substrate (see figure below).
During separation of the two surfaces, a force builds up gradually in the MFPs until one of their two DNA duplexes ruptures, either the target duplex or the reference duplex. After separation, the ratio of ruptured target to reference duplexes is read out via the fluorescence signal of the MFPs on the lower surface and analyzed to obtain the normalized fluorescence (NF). The NF is defined as the ratio of broken reference bonds to the total amount of MFPs that have been under load. Thus the NF is a measure for the relative mechanical stability between target and reference DNA duplexes of a MFP: a higher NF denotes a mechanical stability of the target duplex elevated over that of the reference duplex. The difference in NF reflects a significant difference in mean rupture force between nDNA, mC-1c-DNA and mC-3-DNA.
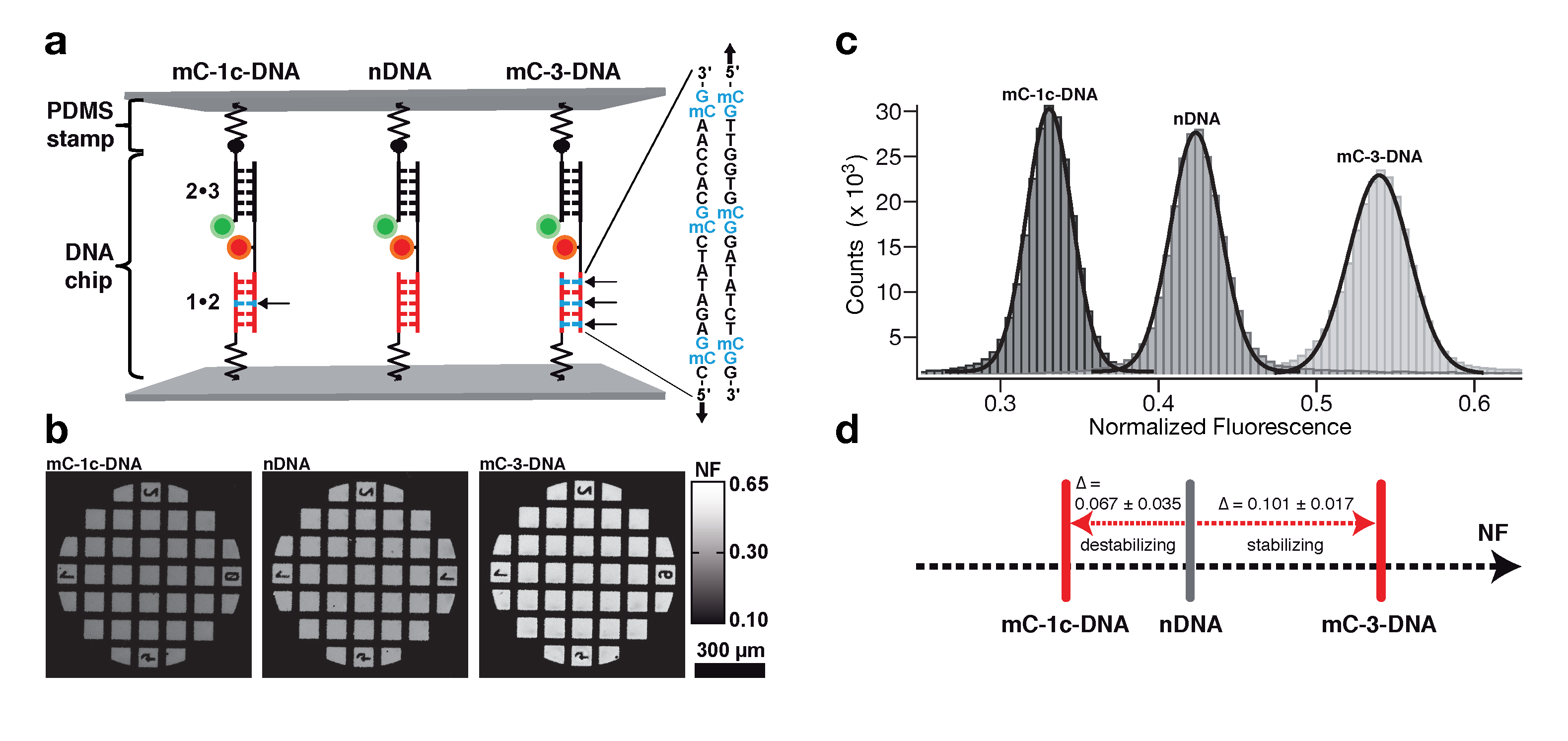
Molecular force assay
Determination of potential width and dissociation constant. To further investigate the differences in strand separation of methylated and non-methylated DNA, single molecule force spectroscopy rupturing the DNA double-strand was performed by AFM. In all experiments one single-stranded DNA (ssDNA) was bound with a poly-ethyleneglycol (PEG) spacer to the cantilever tip of the AFM and a second ssDNA was immobilized with a PEG spacer on a glass substrate. While the tip of the AFM approached the surface, the ssDNA on the tip and the ssDNA on the glass substrate could form a 20 bp duplex. The tip was then retracted from the surface and the DNA duplex was loaded with an increasing force until it finally ruptured (see figure below). The same sequence of the DNA duplex was used as in the MFA experiments with zero (nDNA), one (mC-1c-DNA) or three (mC-3-DNA) 5'-methylcytosines (mC) per strand. Complementing the MFA experiments, the AFM experiments reveal an elevated mechanical stability for mC-3-DNA and a decreased stability for mC-1c-DNA in comparison to nDNA.
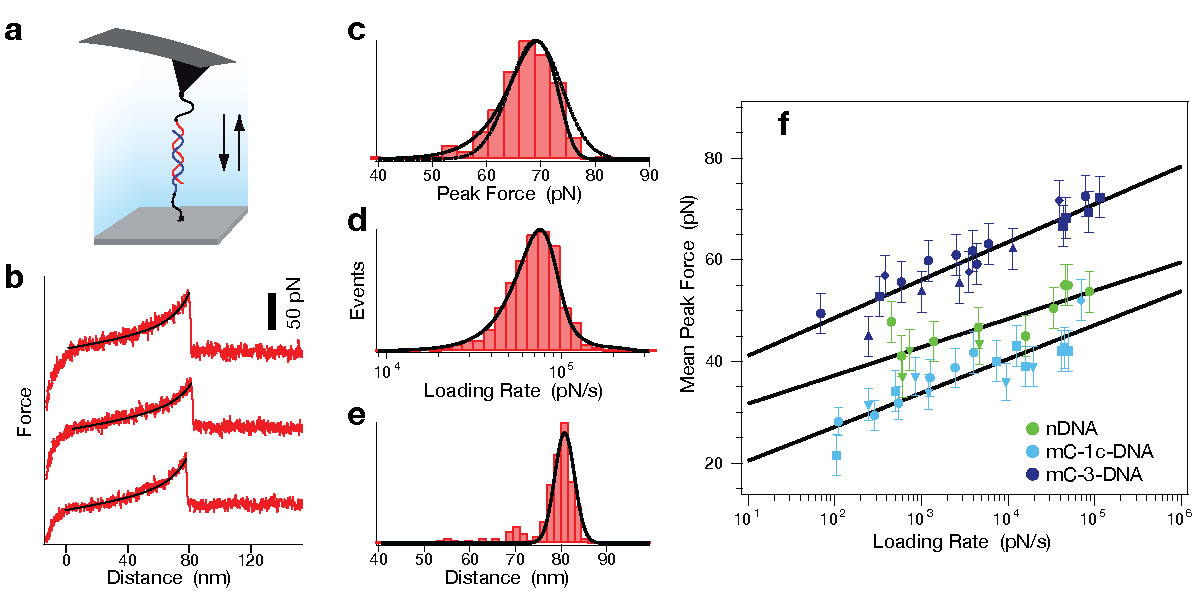
Atomic force microscopy
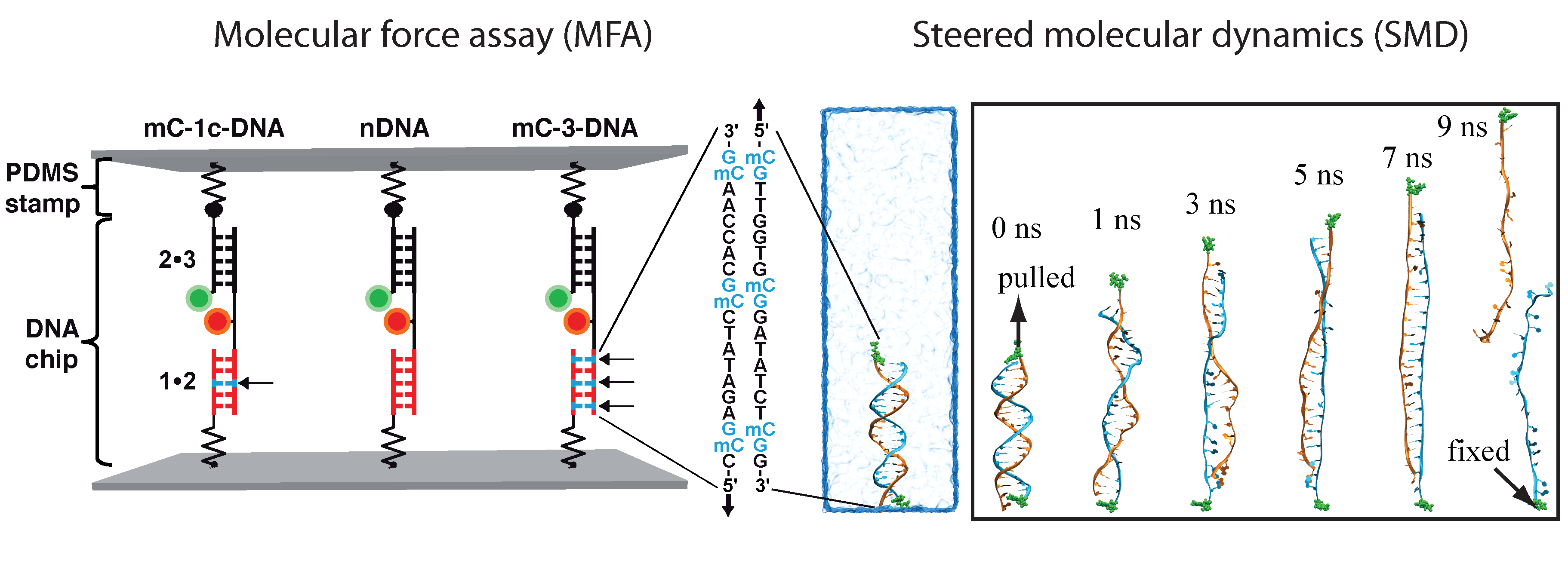
Simulation system
MD simulations of DNA strand separation. In our simulations, non-methylated DNA (nDNA), center-methylated DNA (cDNA) and fully-methylated DNA (fDNA) were subjected to a stretching force directed along the helical axis. Figure below shows force-extension curves for nDNA, cDNA and fDNA stretched with a pulling velocity of 1 Å/ns. The force-extension profile exhibits in each case a clear peak followed by a rapid force decrease. The force-extension curve of cDNA exhibits a minor and a major force peak. Examination of the respective simulation trajectory revealed that some flipped-out bases of the two already separated strands of cDNA stacked on each other after initial, partial separation (minor peak). As a result, a stronger force (major peak) was needed to completely separate the DNA strands. Before DNA reaches an extension of 13.8 nm, the force-extension curves of fDNA, cDNA and nDNA are indistinguishable. However, upon further extension the force needed to extend methylated DNA is larger than the force needed to extend non-methylated DNA, indicating that methylation affects the late-stage of force-induced DNA strand separation.
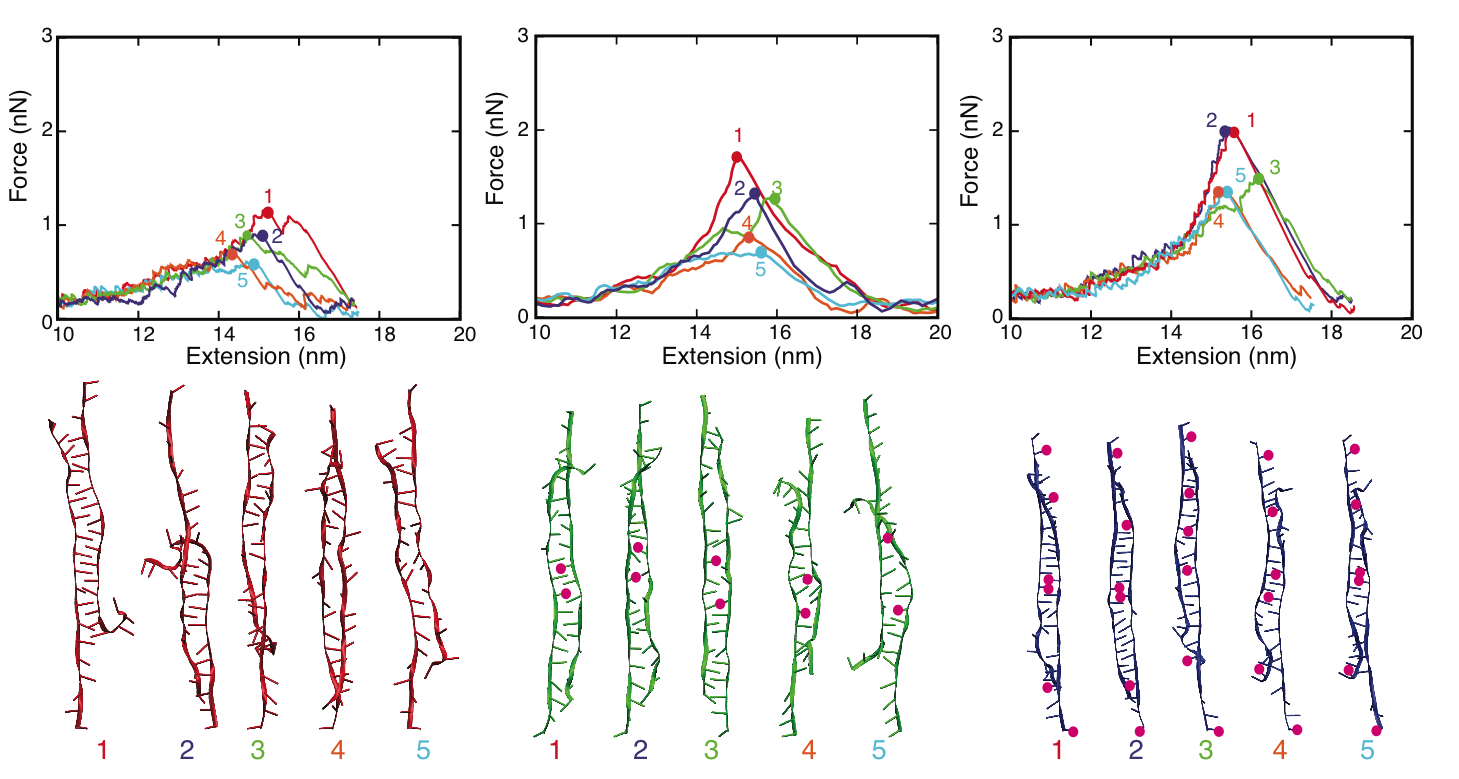
MD simulations
We characterized the strand separation pathway through the 5' end - 5' end distance (extension), stacking energy of DNA bases, and intermediate DNA geometries. Because of thermal fluctuation, some bases keep floating out of the zipper-like packing, and leave holes in the zipper, referred to as ``bubbles". The amount of developed bubbles influences the stability of DNA against strand separation. As can be seen in the figure below, the density of bubbles, on average, is lower for DNA with more methylation sites, which is consistent with the result that fDNA requires a stronger rupture force than nDNA in our simulations.
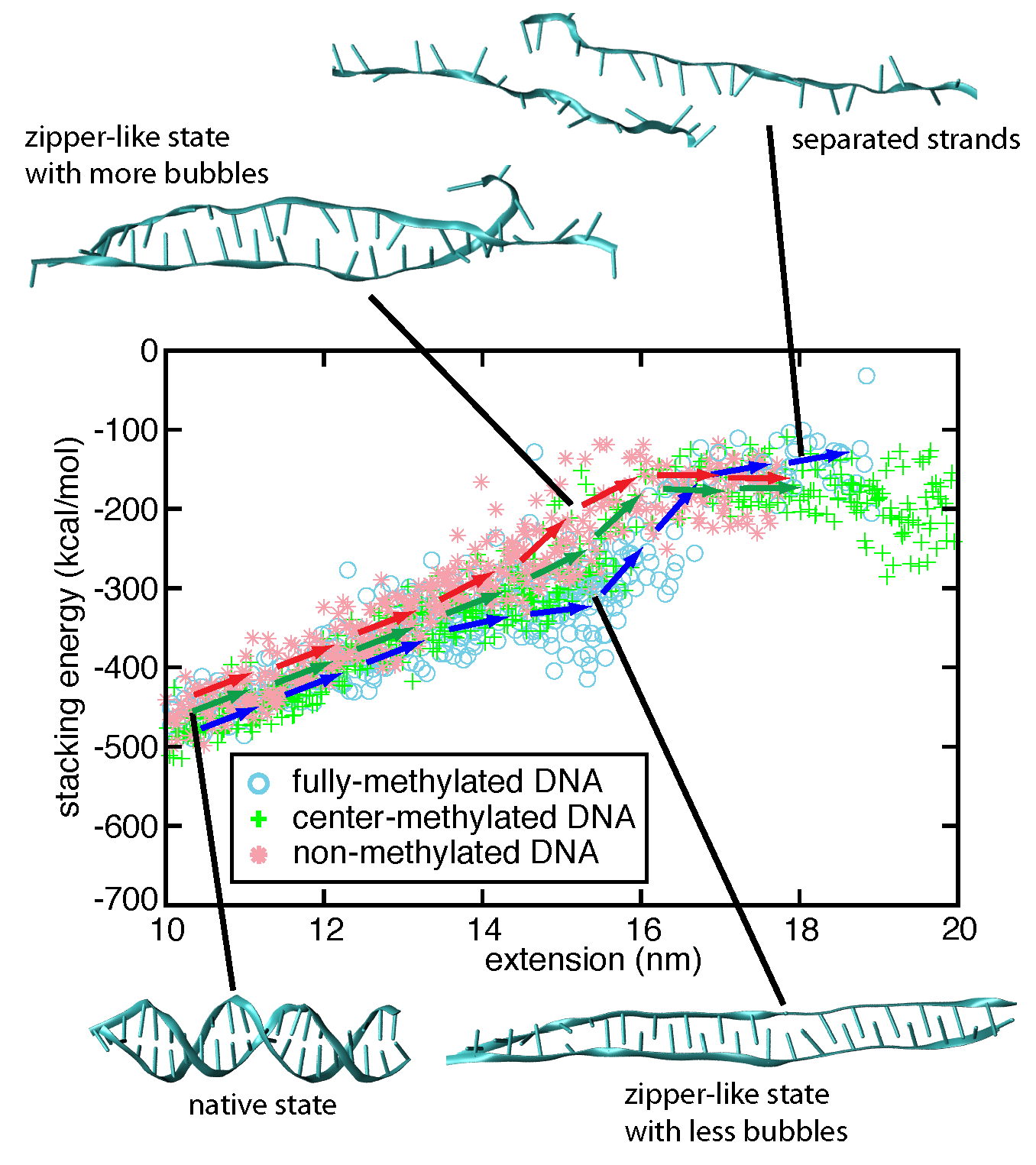
Strand separation pathway of stretched DNA
To investigate the strand separation of nDNA and fDNA under conditions closer to the experimental loading force, we carried out SMD simulations at 200 pN constant force on two DNAs for 90 ns. The figure below shows that under 200 pN stretching, nDNA and fDNA extend along the backbone axis as the DNA increases over 90 ns its extension values. Monitoring stacking energy and length of DNA, one can distinguish two different conformational spaces occupied by nDNA and fDNA. Consistent with the strand separation pathways obtained from fast-pulling (10 Å/ns) SMD simulations, fDNA remains more compact than nDNA does. From the conformations shown in the inset of Figure below, one can see that zipper-like fDNA develops less bubbles than zipper-like nDNA does.
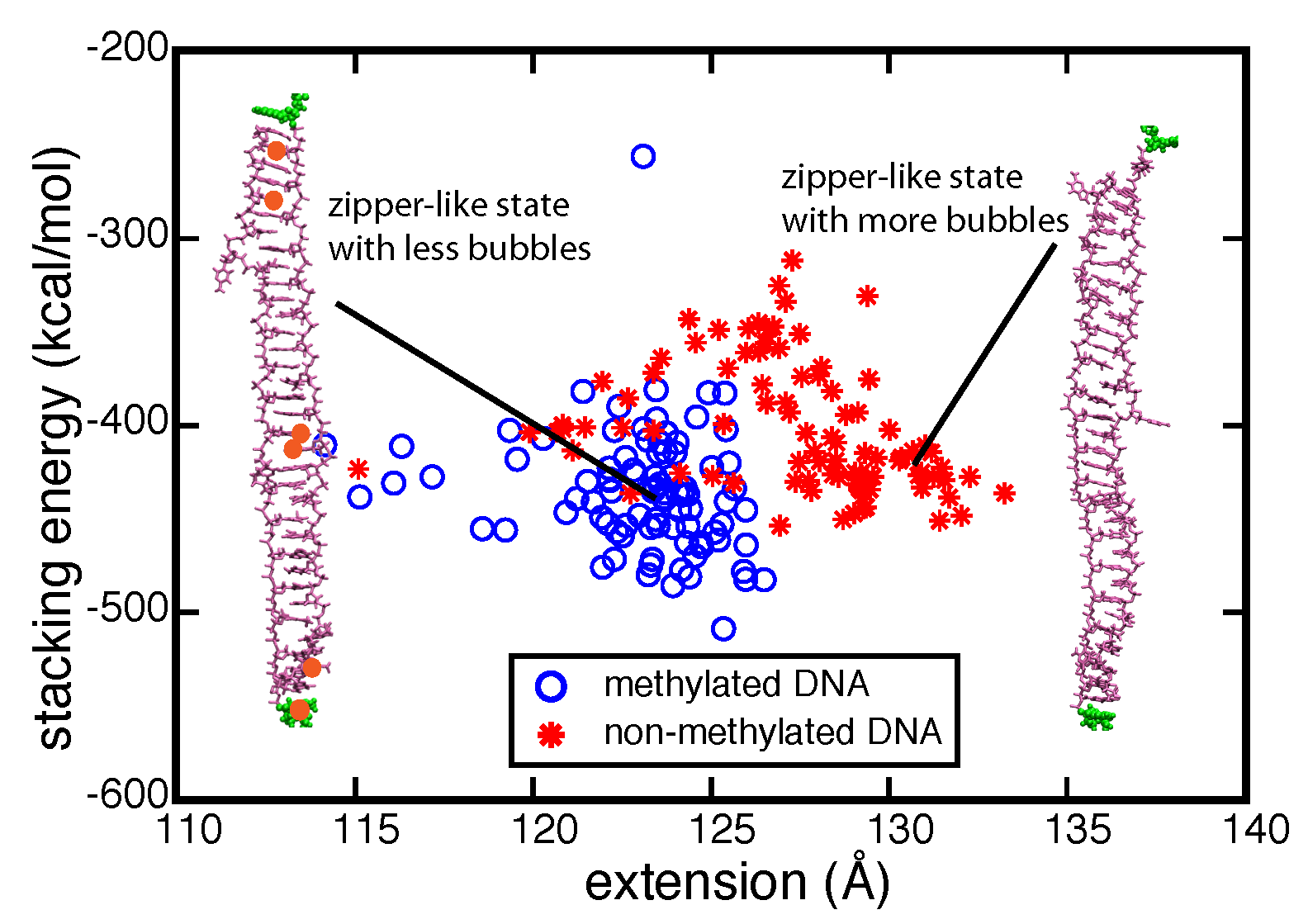
Stacking energy vs. extension data from two 90-ns-long constant force simulations pulling DNA
DNA hydroxymethylation affects strand separation
Molecular force assay setup. Each MFP is comprised of DNA strands 1, 2 and 3, which form two DNA duplexes that are coupled in series. DNA strand 1 is anchored to the substrate (lower surface) and strand 3 is modified with a biotin for coupling to streptavidin on the PDMS-stamp (upper surface). The target duplex exhibits three different variants with none (nDNA), one (hmC-1-DNA) or three (hmC-3-DNA) 5-hydroxymethylated CpG steps, while the reference duplex is the same for all three cases. Furthermore, DNA strand 2 carries a Cy5 (red) and strand 3 a Cy3 (green) fluorescent marker. Depending on the sequences of the DNA strands, the DNA duplexes are oriented such that they are loaded in shear or zipper geometry as shown in (a) and (b).
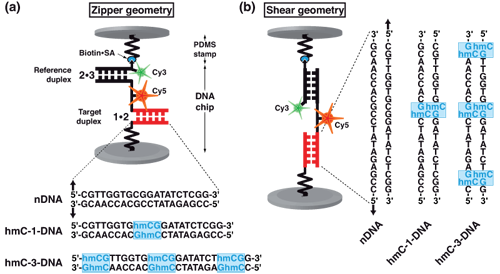
Schematic representation of molecular force probe.
Molecular force assay measurements. Representative results for a typical experiment in zipper and shear geometry are shown in figure below. The Gaussian fits of the histograms of the NF-images result in the mean values and standard deviations, as shown in (a) zipper geometry and (b) shear geometry. The MFA experiments prove a significant influence of hydroxymethylcytosine on the mechanical properties of DNA. The strength of the influence on the mechanical stability depends on the direction of the applied force (zipper versus shear geometry). In zipper geometry we found that the mechanical stability increases with the number of hmC bases. In shear geometry hmC-1-DNA exhibits a lower mechanical stability than nDNA, and hmC-3-DNA a higher stability. Our results indicate that cytosine hydroxymethylation of DNA can both enhance and also decrease the propensity for strand separation. All reported cases show a significant change in mechanical stability of the DNA.
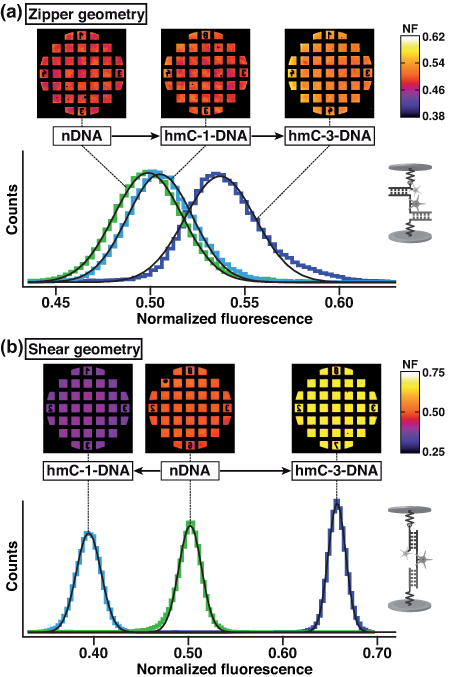
Normalized fluorescence and histograms of one MFA experiment.
Molecular dynamics simulations. In order to demonstrate at the atomic level how hydroxymethylation affects DNA strand separation, SMD simulations were performed to stretch DNA with the same sequence and hydroxymethylation patterns as used in experiments.
When dsDNA is stretched in zipper geometry, the DNA helix unwinds and the base pairs break one by one, forming eventually two extended single-stranded DNA molecules, as shown in figure below (a). In all unzipping simulations, the frequency of appearance of force peaks is close to the frequency of base pair breaking, indicating that almost every base pair breaking event gives rise to a small force peak. After averaging all force values over time, we obtained mean rupture forces for nDNA, hmC-1- DNA and hmC-3-DNA. Consistent with MFA results, hmC-3-DNA exhibits strongest mechanical stability and requires the strongest rupture force.
When dsDNA is stretched in shear geometry, DNA strand separation in two steps. In the first step, DNA elongates and unwinds as can be seen in the 30 ns and 60 ns snapshots in (d). In the meantime, DNA Watson-Crick base pairs begin to break and bases of the two separate strands begin to stack on top of each other, forming the so-called zipper-like DNA, as seen in the 90 ns snapshot in (d). The force increases slowly during the first step (see (e)). In the second step, the acting force increases rapidly. When the pulling force reaches a certain peak value, the two strands of DNA separate (see the 115 ns snapshot in (d)), after which moment the force drops very rapidly to around zero, as seen in (e). Accordingly, a typical rupture force profile for shearing DNA exhibits a peak rupture force, reflecting the DNA stability against strand separation by shearing. In general, hmC-3-DNA assumes a more ordered DNA form with intercalated bases (less bubbles inside the DNA zipper and more compact ends) than do hmC-1-DNA and nDNA; hmC-1-DNA assumes the most disordered conformations, which reduces its mean peak rupture force. Similar to the findings in a previous study on effects of methylation on strand separation of methylated DNA, hydroxymethylation also affects bubble formation during DNA stretching, which controls the propensity for strand separation.
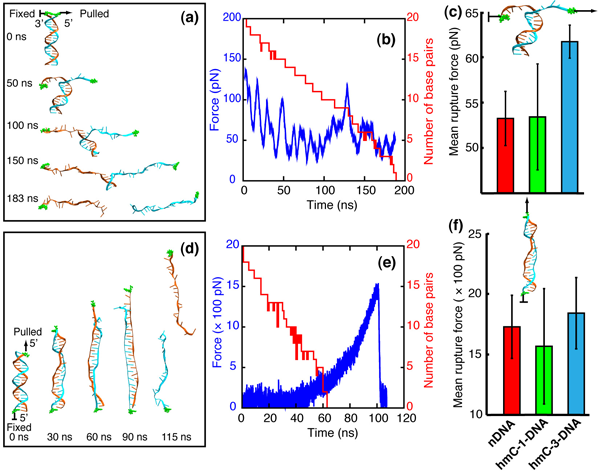
MD simulations.
Summary. We demonstrated through experiment and simulation a pronounced effect of methylation and hydroxymethylation on the propensity of DNA strand separation. Even though we identified this effect, we could not uncover fully the associated molecular mechanism. Therefore, further investigations are needed. The effect of hydroxymethylation exceeds in strength what was previously demonstrated for the effect of methylation. Our study reveals that methylation and hydroxymethylation could regulate gene expression through altering mechanical properties of DNA.
Methyl-CpG binding domain (MBD) proteins recognize methylated DNA
MBD protein family. At present, five proteins with mCpG-binding motifs have been identified in the MBD family, namely MBD1, MBD2, MBD3, MBD4 and MeCP2. All MBD proteins share a similar MBD of about 75 amino-acid residues. Sequence alignment of MBD proteins reveals several highly conserved residues on the interface between MBD and methylated DNA (mDNA). Figure below shows that the most conserved region in the primary structure of the MBD is located between residues ARG22 and LYS46, including loop L1, strand β3, strand β4 and loop L2. The conserved residues of the MBD form the interfacial surface contacting mDNA and are responsible for recognition of the methyl-CpG steps.
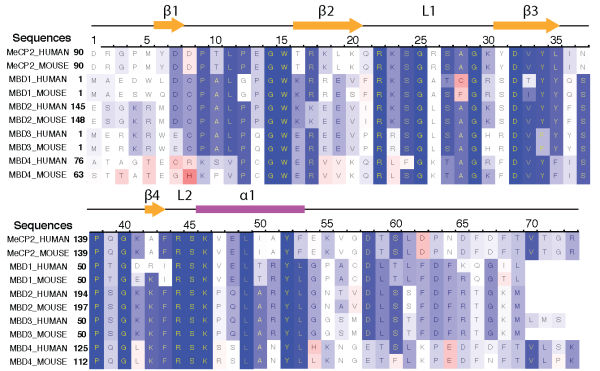
Sequence alignment of five methyl-CpG binding domain (MBD) proteins from human and mouse.
MBD proteins bound to mDNA. The NMR structures of MBD1, MBD2 and the X-ray structure of MeCP2 proteins bound to the mDNA provide useful information on how residues in the MBD contact mDNA. Figure below illustrates the MBD interaction with the mCpG steps in the major groove of mDNA. The methyl groups of methyl-cytosine are buried in the mDNA-MBD interface. The principal mCpG binding interface composed of hydrophilic and hydrophobic residues involves two types of interaction with mCpG steps: the polar atoms in MBD form hydrogen bonds with the polar atoms in the DNA, while the non-polar atoms in MBD form a hydrophobic patch around the methyl groups.
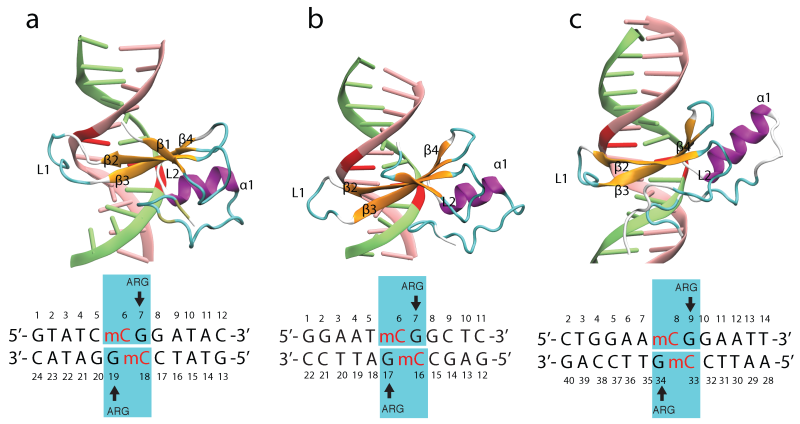
MBD proteins binding to mDNA. Shown are the structures of (a) MBD1-mDNA, (b) MBD2-mDNA and (c) MeCP2-mDNA complexes.
Molecular dynamics simulation. Structures of MBD1-mDNA, MBD2-mDNA and MeCP2-mDNA complexes reveal that the conserved residues ARG22 and ARG44 form hydrogen bonds with guanines in the mCpG steps. To investigate the role of ARG22 and ARG44 in recognizing the mCpG steps, we conducted a 100-ns molecular dynamics simulation of the NMR structure of the human MBD1-DNA complex. Simulations showed that the spatial configurations of mCYT106-ARG22-GUA107 and mCYT118- ARG44-GUA119 adopt a so-called stair motif. Similar stair motifs are formed and remain stable at the MBD2-mDNA and MeCP2-mDNA interface.
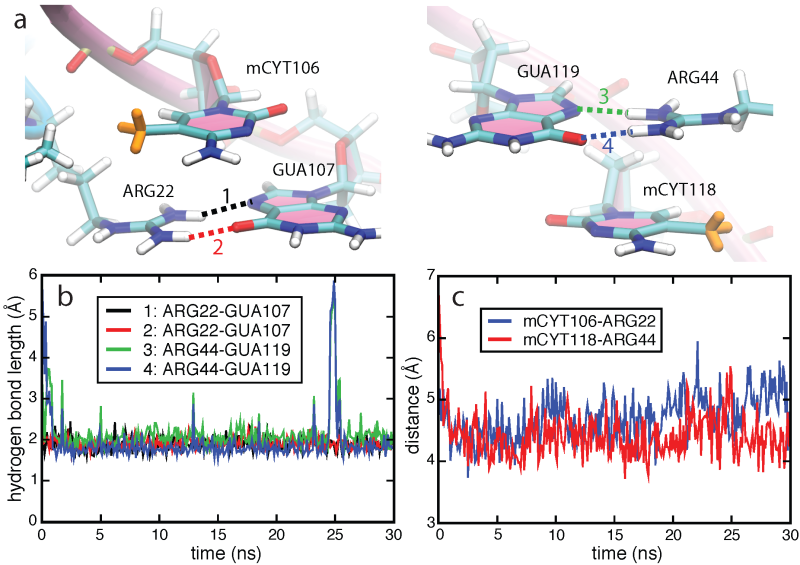
Stair motifs in the interface of MBD1-mDNA.
Mutational studies in which mCYT106 and mCYT118 were substituted by nCYT106 and nCYT118 (nCYT denotes non-methylated cytosine) revealed that MBD1 does not bind to non-methylated DNA, indicating that the 5-methyl groups on the methyl-cytosines are critical for the formation of the stair motif in MBD-mDNA binding interfaces. The influence of methylation on the stability of the stair motif was investigated through a 30-ns MD simulation for the MBD1-nDNA complex, where the mCYT106 and mCYT118 nucleotides were mutated into two non-methylated cytosines. Our resutls showed that the interactions between ARG and mCYT are more stable than those between ARG and nCYT, as methylation increases the contact area between mDNA and the MBD.
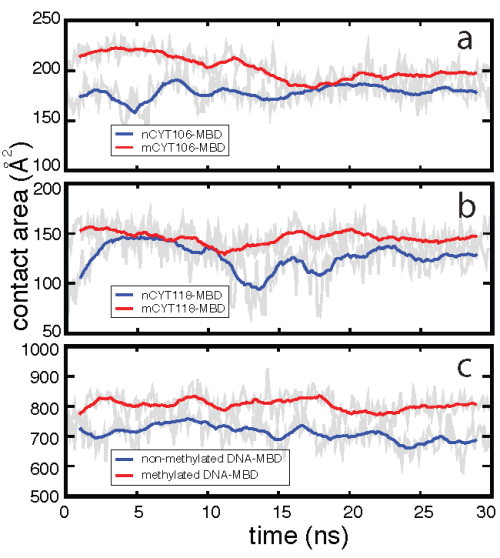
Effect of methylation on the MBD1-DNA contact area.
Ab initio quantum chemistry calculations. We performed quantum chemistry calculations of the interaction energy in the mCYT-ARG-GUA and nCYT-ARG-GUA stair motifs in order to determine the impact of the methyl group on the stair motif stabilization. QM calculations show a stabilization effect of cytosine methylation on the studied system, see figure below. The blue line (squares) shows the interaction energy calculated for the mCYT106-ARG22-GUA107 stair motif, the red line (triangles) shows the interaction energy calculated for the mCYT118-ARG44-GUA119 stair motif, while the green line (dots) shows the combined energy difference.
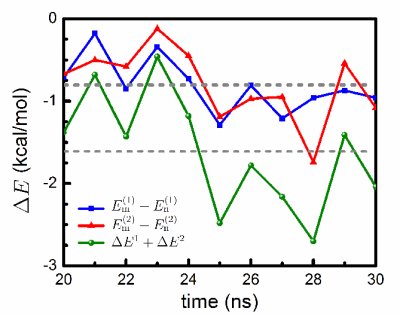
Influence of cytosine methylation on MBD1 binding energy.
Free-energy contributions of DNA methylation to MBD1-DNA complex stabilization. The protein-DNA binding affinity is affected by different interactions. In order to account for the total effect of methylation on the stability of the MBD-DNA complex, we performed FEP calculations to characterize the free-energy contribution of the methyl group to the MBD-DNA complex. The BAR (see Methods) free-energy difference for the free DNA was determined to be ΔG1 = 54.9 ± 0.0 kcal/mol, with -0.12 kcal/mol hysteresis between forward and backward transition; for the MBD-DNA complex, the free-energy difference was determined to be ΔG2 = 53.7 ± 0.1 kcal/mol, with 0.2 kcal/mol hysteresis between forward and backward transition. Following thermodynamic cycle shown below, one can obtain ΔΔG = -1.2 ± 0.1 kcal/mol, hence, indicating that methylation makes a favorable contribution to the binding free energy.
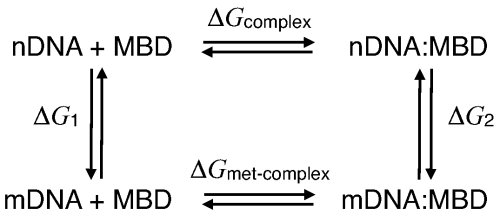
Thermodynamic cycle describing DNA methylation in the DNA-MBD complex.
Publications
-
Effects of cytosine hydroxymethylation on DNA strand separation Philip M.D. Severin*, Xueqing Zou*, Hermann E. Gaub, and Klaus Schulten Biophysical Journal, In press
-
Recognition of methylated DNA through methyl-CpG binding domain proteins Xueqing Zou, Wen Ma, Ilia Solov'yov, Christophe Chipot and Klaus Schulten Nucleic Acids Research, 40:2747-2758,2012
-
Cytosine methylation alters DNA mechanical properties Philip M.D. Severin*, Xueqing Zou*, Hermann E. Gaub, and Klaus Schulten Nucleic Acids Research, 39:8740-8751, 2011
-
Nanoelectromechanics of Methylated DNA in a Synthetic Nanopore U. Mirsaidov, W. Timp, X. Zou, V. Dimitrov, K. Schulten, A. P. Feinberg, and G. Timp Biophysical Journal, 96:L32-L34, 2009
* Equal contribution authors
Investigators
Simulations ExperimentRelated TCB Group Projects
Page created and maintained by Xueqing Zou.


