Wen Ma

Update (Aug. 2018): currently I'm a postdoc researcher in Andy McCammon's group at UCSD.
For latest updates, check out my Google Scholar
Contact Information
- Ph.D. in Biophysics and Computational Biology, University of Illinois at Urbana-Champaign, Aug. 2017
- B.S. in Physics, Peking University, July 2010
- Graduate College travel grant for Gordon Research Conference - Proteins, 2015
- Travel award, Biophysical Society 2015 annual meeting
- Award for outstanding undergraduate research, Peking University, Nov. 2009
- Wusi fellowship, Peking University, 2007~2009
- Talk: "Molecular Dynamics Study of Directional Translocation and Subunit Coordination in a Hexameric Helicase Motor." NSF's Physics of Living Systems (PoLS) annual meeting, Yale University, July 2012.
- Talk: "Computational Study of RNA Translocation in a Hexameric Helicase." 57th Biophysical Society annual meeting, Philadelphia, February 2013.
- Talk: "Study of Conformational Transitions of Molecular Machines Using the String Method." Annual meeting of the Membrane Protein Structural Dynamics Consortium, Chicago, May 2013.
- Invited Talk: "Physical Mechanism of Rho Hexameric Helicase." Center for the Physics of Living Cells (CPLC) symposium, University of Illinois, October 2013.
- Invited Talk: "A Lesson from Nature: Teamwork Is the Key to Success.", the Illinois Biophysics Society, Urbana, Jan. 2015.
- Talk: "Protein Unfolding by a Ring-shaped Unfoldase." Seminar, University of California, Berkeley, Aug. 2016.
- "Force Generation Mechanism of Molecular Motors." Poster section for NSF site visit to CPLC, University of Illinois, April 2013.
- "RNA Translocation Coupled to Large-Scale Conformational Transitions of a Hexameric Helicase." 59th Biophysical Society annual meeting, Baltimore, February 2015.
- "Mechanism of Substrate Translocation by a Ring-Shaped ATPase Motor at Millisecond Resolution." Gordon Research Conference: Proteins, Holderness, June 2015.
Postdoctoral Scholar, Department of Chemistry and Pharmacology, UCSD
Urey Hall 4234, 9500 Gilman Drive MC 0340, La Jolla, CA 92093
Email: w1ma@ucsd.edu or wenma@ks.uiuc.edu
Education
Below not up-to-date
Research accomplishments
The underlying theme of my research is to characterize ways in which laws of physics yield biomolecular function over a wide range of temporal and spatial scales. Specifically, my work focused on developing multiscale simulation and analysis methods to extend all-atom simulations to biologically relevant timescales [1-7]. These methods enabled me to study molecular machines that participate in many key cellular processes, including DNA repair, gene expression, protein transport, and protein degradation.
1. Advanced sampling techniques
Molecular dynamics (MD) simulations, though capable of providing atomic details, are limited in the study of molecular machines owing to the challenge that these systems function on a millisecond timescale which, for a long time, could not be easily covered computationally. One of my most important development involves advanced sampling techniques based on the so-called transition path theory, to study the mechanochemical energy transduction in molecular machines (for theoretical details see Supporting Information in Ref.4). To obtain the transition paths as well as the related free energy landscape and kinetics, I have implemented the string method with swarms of trajectories combined with the milestoning technique with NAMD. Recent progress included further development of the milestoning method enabled by a multiplex replica algorithm, which allows very efficient allocation of the computational resource among a large number of independent MD trajectories [in preparation]. Download link: the packages can be found here. The link includes three packages (compatible with the use with NAMD): the string method with swarms of trajectories, milestoning (with reflective boundary), and a multiplexed replica protocol (e.g. for milestoning).
2. Mechanochemical energy transduction and allostery in molecular machines
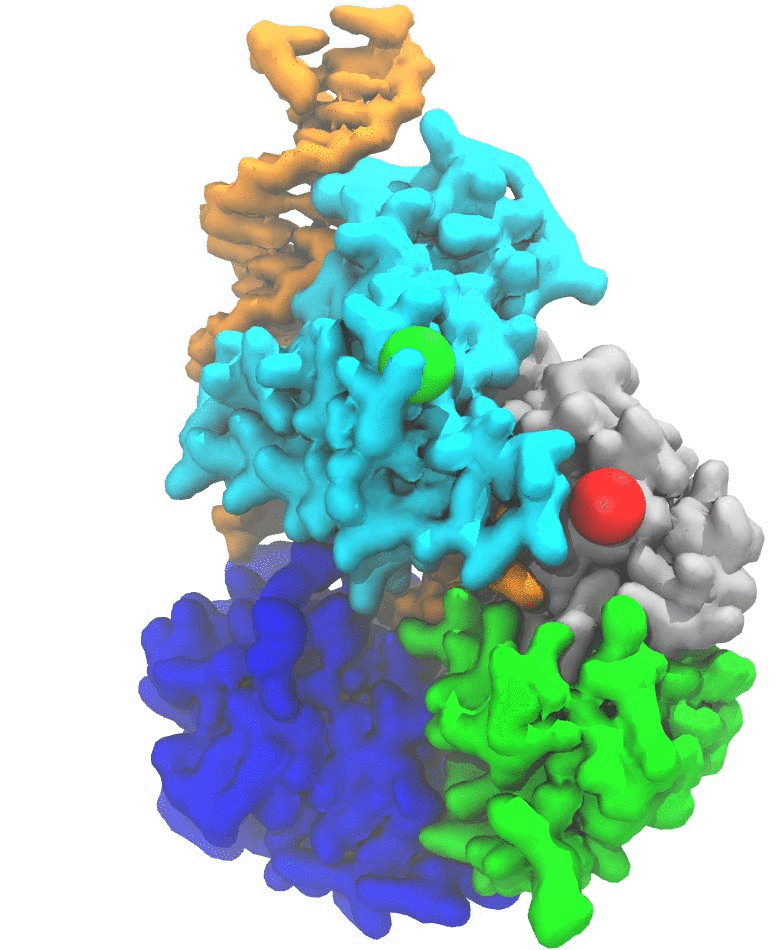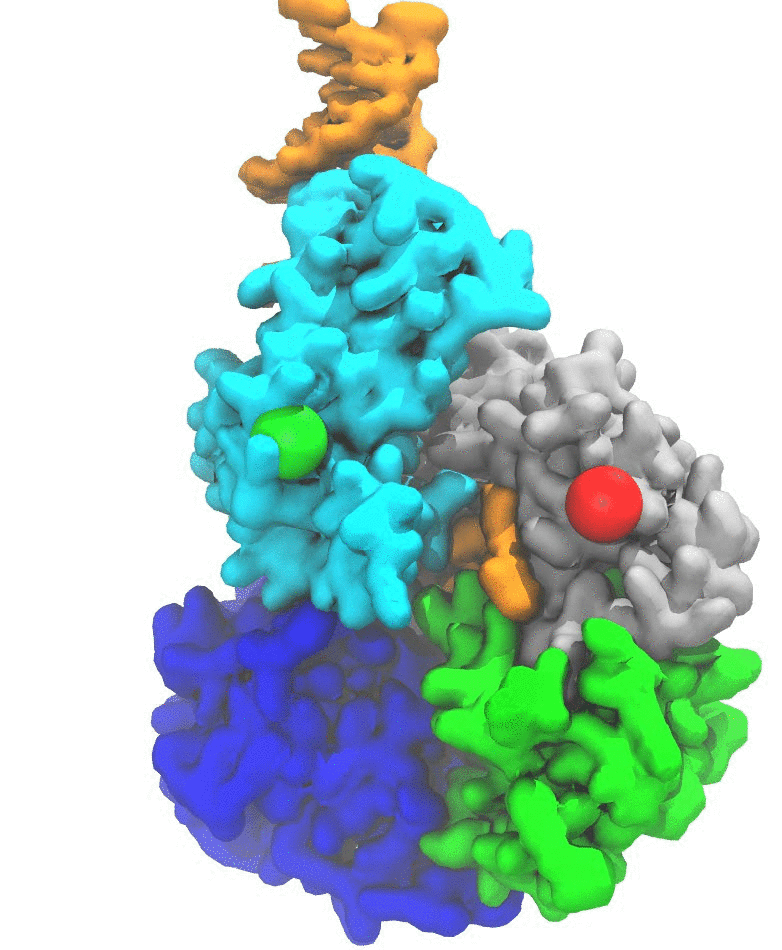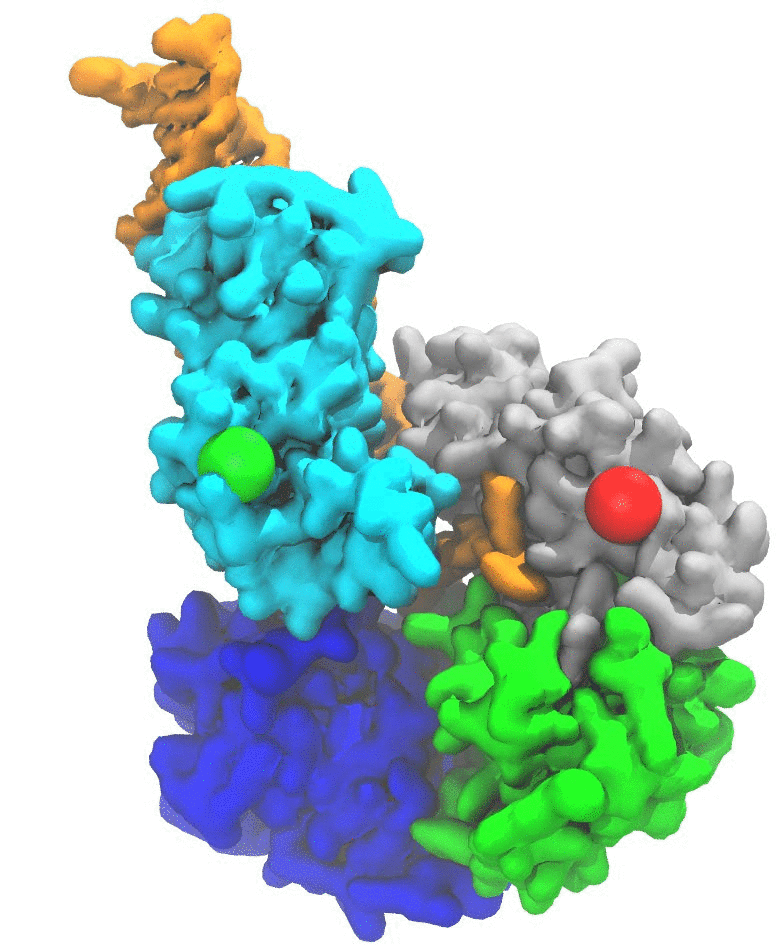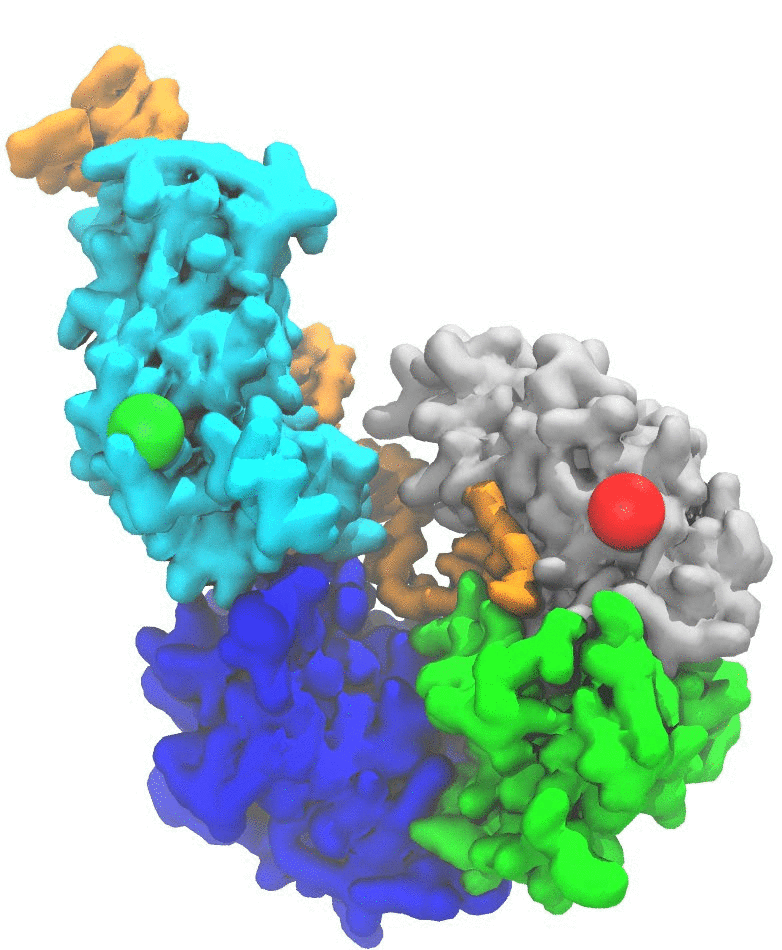
(2a) RNA translocation by Rho helicase. MD simulations combined with transition path theory enabled me to study the highly coordinated motion of Rho hexameric helicase while it unidirectionally translocates RNA (Fig. 1) [4]. Our simulations showed that the release of hydrolysis product (ADP+Pi) triggers the force-generating process of Rho through a 0.1 millisecond-long conformational transition, of which a similar timescale was also seen in experiments. The study not only revealed in new detail the mechanism employed by ring-shaped ATPase motors, for example, the use of loosely bound and tightly bound hydrolysis reactant and product states to coordinate motor action, but also provided an effective approach to identify allosteric sites of multimeric enzymes in general. See more information in TCBG news, and the research page, as well as a youtube video made by me.
Inspired by the allosteric network in Rho revealed in [4], we started a collaboration with the crystallographers to investigate how a universal adapter protein NusG regulates the function of Rho - transcription termination. Our work [2] demonstrated that the universally conserved factor NusG harnesses the activity of the ring-shaped motor Rho through allostery, and established a new mechanism for sequence-independent transcription termination.|
|
|
|
Fig. 1. Mechanism of RNA translocation by Rho helicase. ADP+Pi release triggers Rho transitioning from an initial state I to a final state F. The initial ligand binding states, after a 60° clockwise rotation around the z-axis, are the same as those of the final state. In going from I to F, a molecule of RNA is propelled through Rho's central pore. The schematic free energy landscape, governing the transition, is shown in the middle. |
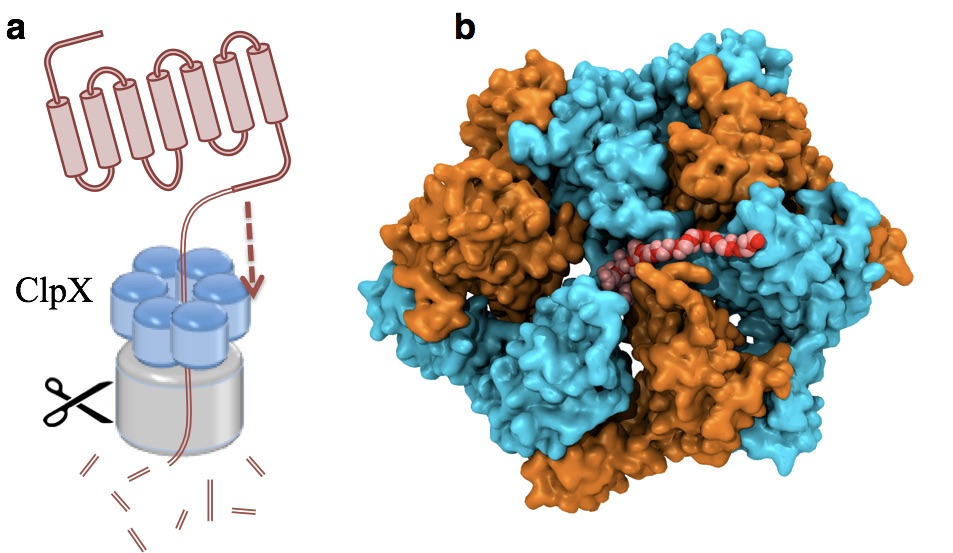
(2b) Protein unfolding by ClpX unfoldase. Following the footsteps of the study on Rho, we applied the pathway sampling techniques to study how an unfoldase ClpX uses its central pore loops to translocate polypeptides during its mechanochemical cycle (Fig. 2). The ClpX ATPase belongs to the large family of AAA+ unfoldases, which function in isolation or associated with compartmental peptidases to remove damaged or misfolded polypeptides and turn over regulatory proteins that control numerous vital processes in all cells. Our collaborator's experiments implied that phosphate release after ATP hydrolysis drives ClpX conformational changes, and that the translocation step size is determined by the number of hydrolyzing ClpX subunits. We have characterized in atomic detail how phosphate release triggers the long-timescale conformational changes representing a basic power stroke for mechanical substrate processing.
|
|
|
|
Fig. 2. ClpX unfolds and translocates a protein by pulling its terminus through its central pore. Such a mechanical process is powered by ATP hydrolysis via conformational changes of the ClpX hexamer. (a) A schematic representation of ClpX unfolding a protein. (b) An intermediate state during polypeptide translocation by ClpX. |
3. Characterize protein sequence-structure-function relationships
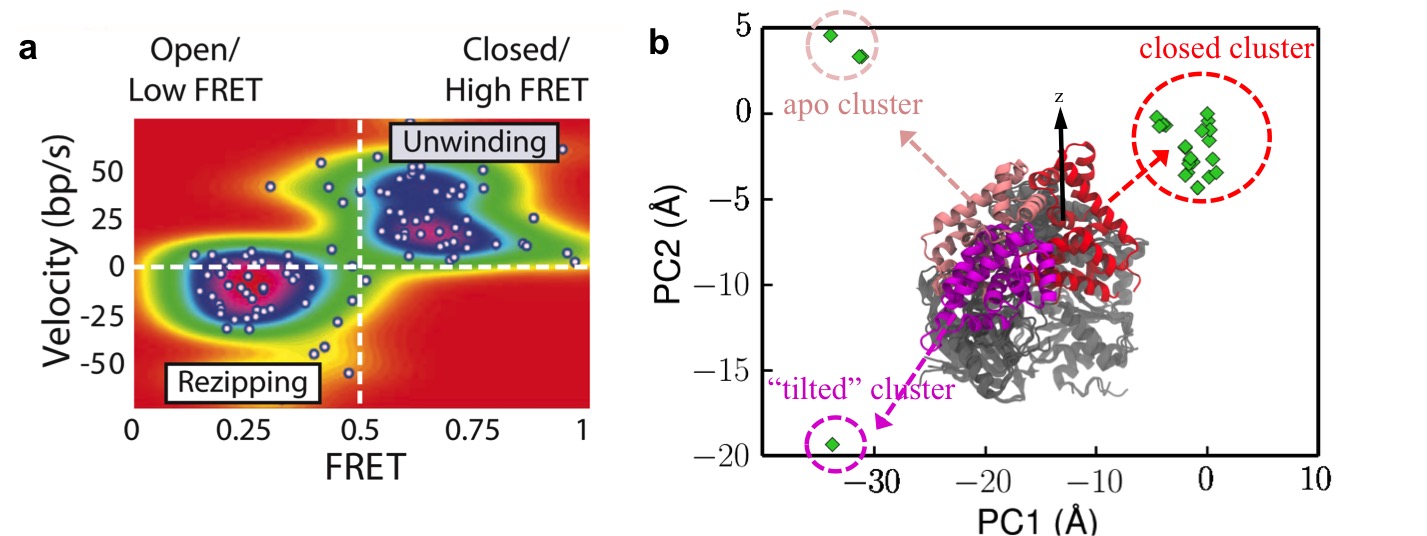
An interesting question in biophysics is how to bridge the information from protein sequence with their structure and function. Here we investigated an exemplary DNA-processing enzyme UvrD helicase, which plays key roles in DNA replication and repair, by unwinding nucleic acid strands. Collaborating with Prof. Yann Chemla (University of Illinois), who used optical tweezer combined with FRET to study the single-molecule behavior of UvrD (Fig. 3a), we characterized the mechanism of which UvrD changes its conformation to alter its function from unwinding to rezipping dsDNA, by applying advanced sampling simulations[1] (movie). We also predicted a key metastable state (tilted state) for the functional switch of UvrD. Our simulations were enabled by structural bioinformatics combined with principal component analysis (Fig. 3b).
|
|
|
|
Fig. 3. (a) Population distribution calculated using unwinding velocity and FRET signals (adapted from Fig. 3D of Comstock, ..., Chemla, Science 348: 352 (2015)). The map shows a strong correlation between UvrD activity and UvrD conformation. (b) Projection of surveyed crystal structures onto the first two modes from principal component analysis, which reveals three different structural clusters. One of them is unexpected and found in the present study to be crucial for UvrD functional switching. |
4. Multiscale modeling
My endeavors also involved developing theoretical models to understand simulation or experimental data from my collaborators. In the first year of my Ph.D. study, I formulated a statistical model to study how flagellin monomers are transported to grow the bacterial flagellum [7] (an hours-long process), based on the parameters derived from MD simulations.
Later on, employing the free energy perturbation method, I was able to reveal the impact of cytosine methylation on the recognition of DNA binding proteins, illustrating the importance of free energy landscapes in understanding epigenetic modifications [6].
Recently collaborating with my colleagues, we applied a coarse-grained model to characterize the structural ensembles of monomeric alpha-synuclein, in which a beta-hairpin was formed and expected to be crucial for alpha-synuclein aggregation [5]. This simulation result was later dramatically confirmed by an experimental study (Salveson et.al., JACS, 2016, 138: 4458).
During my undergraduate study, I used computational modeling (Poisson-Nernst-Planck theory and related numerical PDEs) to help experimentalists improve the energy conversion efficiency of their battery device powered by concentration gradient in an ion-selective nanofluidic channel [8,9].
Publications
[1] W. Ma , K. Whitley, Y. Chemla, Z. Schulten and K. Schulten. Free energy simulations reveal molecular mechanism for functional switch of a DNA helicase. eLife, 7, e34186, 2018.
[2] M. Lawson, W. Ma , M. Bellecourt, I. Artsimovitch, A. Martin, R. Landick, K. Schulten and J. Berger. Mechanism for the regulated control of transcription termination by a universal adapter protein. Mol. Cell., 71(6), 911-922, 2018.
[3] Q Zhang, et al. Designing a Green Fluorogenic Protease Reporter by Flipping a Beta Strand of GFP for Imaging Apoptosis in Animals. Journal of the American Chemical Society, 141(11), 4526-4530, 2019.
[4] W. Ma and K. Schulten. Mechanism of substrate translocation by a ring-shaped ATPase motor at millisecond resolution. Journal of the American Chemical Society, 137(8), 3031-3040, 2015. (SI here)
[5] H. Yu, W. Han, W. Ma , and K. Schulten. Transient β-hairpin formation in α-synuclein monomer revealed by coarse-grained molecular dynamics simulation. J Chem Phys, 143(24):243142, 2015.
[6] X. Zou, W. Ma , I. A. Solov'yov, C. Chipot, and K. Schulten.Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Research, 40(6), 2747-2758, 2012. (Recommended in F1000 Prime)
[7] D. Tanner, W. Ma , Z. Chen, and K. Schulten. Theoretical and computational investigation of flagellin translocation and bacterial flagellum growth. Biophysical Journal, 100(11), 2548-2556, 2011. (SI here)
[8] L. Cao, W. Guo, W. Ma , L. Wang, F. Xia, S. Wang, Y. Wang, L. Jiang, and D. Zhu. Towards understanding the nanofluidic reverse electrodialysis system: Well matched charge selectivity and ionic composition. Energy & Environmental Science, 4(6), 2259-2266, 2011.
[9] W. Guo, L. Cao, J. Xia, F. Nie, W. Ma , J. Xue, Y. Song, D. Zhu, Y. Wang, and L. Jiang. Energy Harvesting with Single-Ion-Selective Nanopores: A Concentration-Gradient-Driven Nanofluidic Power Source. Advanced Functional Materials, 20(8), 1339-1344, 2010.
Awards and Honors
Talks and Lectures
Poster Presentations



{kind=link}