The Dynamics of Protein Translocation
Protein Localization in the Cell
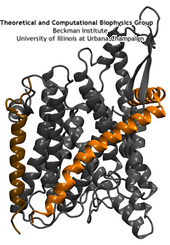 The protein-conducting channel.
The protein-conducting channel.
Proteins are the workhorses of the typical cell. Originally coded for by DNA and composed of a sequence of amino acids, proteins serve a variety of roles including binding other molecules and/or transporting them, serving as 'switches' for cellular activity, catalyzing chemical reactions, and even acting as structural building blocks. It should come as no surprise then that in order to accomplish all these goals, proteins are needed in a variety of locations both inside and outside of the cell. Therefore, a variety of mechanisms have evolved in order to correctly localize proteins, helping to guide them from their point of synthesis (at the ribosome) to their destination. One such set of mechanisms is the secretory pathway.
Also ubiquitous in biological systems are membranes, barriers between the cell and the outside world. Membranes also serve to divide compartments within some cells (those
of eukaryotes). In order for proteins to get to where they are needed inside these compartments or outside the cell, they need a way to cross the membrane without rupturing it at the same
time. The secretory pathway consists in part of a channel for just this purpose: the translocon (
![]()
![]()
![]() ).
).
A Dynamic Channel
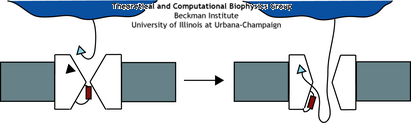 A schematic of the translocation process. The ribosome docks with the translocon where the signal
sequence (triangle) is recognized. This then opens the channel allowing the protein to
go through.
A schematic of the translocation process. The ribosome docks with the translocon where the signal
sequence (triangle) is recognized. This then opens the channel allowing the protein to
go through.
The translocon may be comprised of multiple proteins, but the core component is a heterotrimeric protein known as the Sec61 complex in eukaryotes, SecYEG in bacteria, and
SecYEβ in archaea. Despite their differences, all function in a similar way. The ribosome, the factory for all proteins, docks with the translocon where it begins feeding the new,
still linear, protein into the channel as it's being made, a process known as co-translational translocation (see also our ribosome research page). The channel appears to open up just a few angstroms wide, not enough for
a completely folded protein but enough for a perhaps a helix. To guarantee not just any molecule can cross the channel though, the translocon will only open for proteins displaying the right
'key', in this case, a short specific amino acid leader sequence. This key is recognized by SecY/Sec61 itself, although the manner in which this causes the channel to open is still a
mystery. Originally theorized over 30 years ago, it was the hypothesis of this key and channel combination that earned Günter Blobel the Nobel Prize in Physiology or Medicine in 1999
(![]() ).
).
Once the channel has been opened, some may assume that it is a passive conduit for the nascent
proteins being fed into it. However, this is actually where the channel begins its most complex role, that of deciding
whether to place the protein on the other side of the channel or to insert it into the membrane. While
translocation of soluble proteins across the membrane appears to be straightforward, there are different suggestions for
how segments of protein destined for the membrane insert into it. Some ideas include modification of SecY/Sec61 by the
ribosome
(![]() )
or a thermodynamic partitioning of the hydrophobic transmembrane protein segments between the solvated channel interior
and the hydrophobic membrane core while inside the channel
(
)
or a thermodynamic partitioning of the hydrophobic transmembrane protein segments between the solvated channel interior
and the hydrophobic membrane core while inside the channel
(![]() ).
Regardless, it seems clear that large structural changes are required to open this channel both to membrane as well as to the opposite side.
).
Regardless, it seems clear that large structural changes are required to open this channel both to membrane as well as to the opposite side.
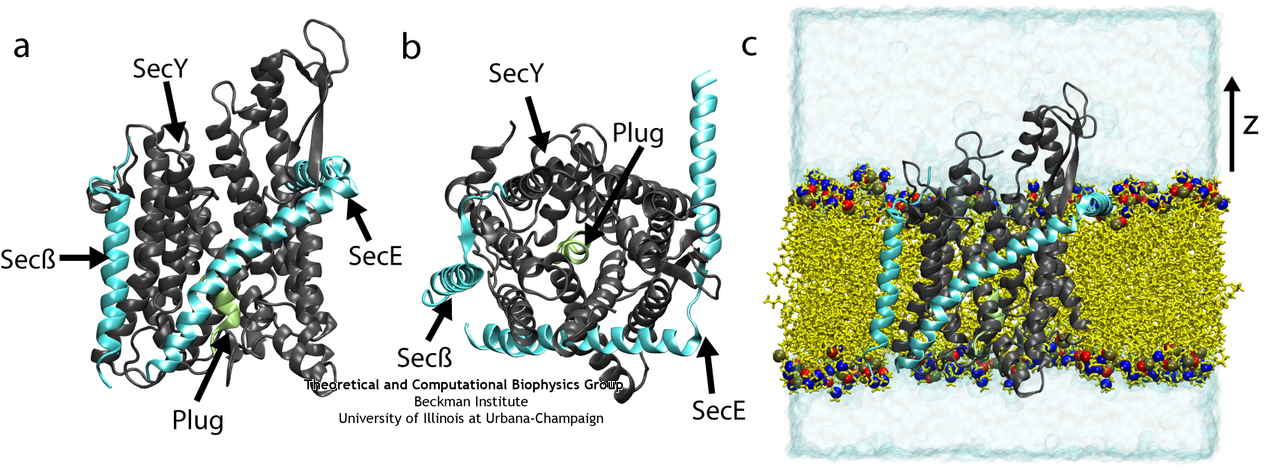
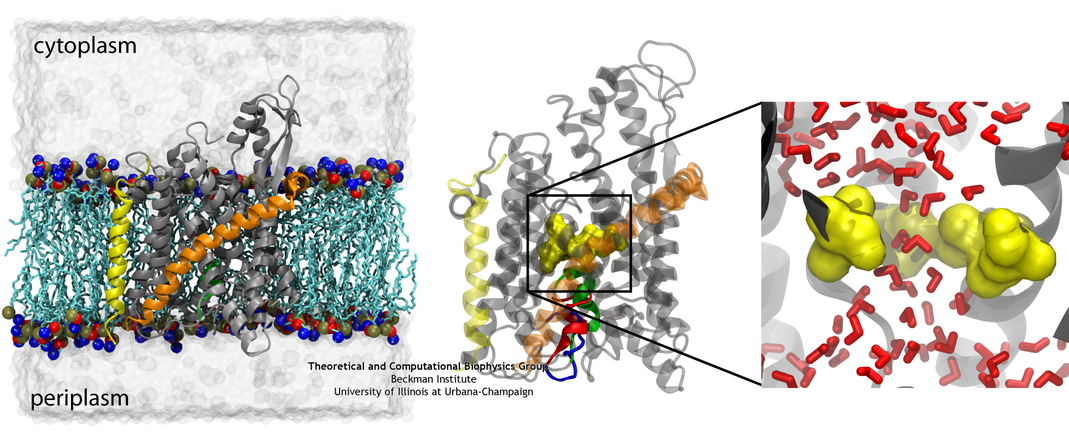
SecYEβ, shown here both from the side and from above and also in its native membrane/water environment (click for a larger view).
Moving Proteins Across the Channel
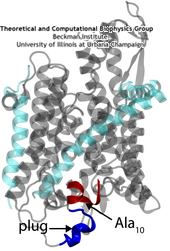 A small polypeptide (protein segment) is crossing the channel and pushing open the plug of SecY.
A small polypeptide (protein segment) is crossing the channel and pushing open the plug of SecY.
The recent availability of a crystal structure of SecYEβ
(![]() )
has brought into focus some aspects of the protein translocation process. Two key structural elements were
identified that help keep the channel closed before and during translocation. First is a 'plug', a small
helix blocking the channel's periplasmic side (opposite of where proteins are fed into the channel).
Second is a 'pore ring', a constriction point at the center of the channel consisting of a few residues from
different helices. Molecular dynamics simulations using the program NAMD have allowed us to characterize
these elements and confirm their hypothesized function (described more thoroughly in a recent paper
)
has brought into focus some aspects of the protein translocation process. Two key structural elements were
identified that help keep the channel closed before and during translocation. First is a 'plug', a small
helix blocking the channel's periplasmic side (opposite of where proteins are fed into the channel).
Second is a 'pore ring', a constriction point at the center of the channel consisting of a few residues from
different helices. Molecular dynamics simulations using the program NAMD have allowed us to characterize
these elements and confirm their hypothesized function (described more thoroughly in a recent paper
![]() ).
During simulated translocation of short polypeptide helices, the plug was seen to leave the channel and
then, after translocation, return partially to its original location. Also, the pore ring blocked water and
ions before translocation; during translocation, it expanded enough to allow the polypeptide segment through
but did not allow any ions through and only a few water molecules.
).
During simulated translocation of short polypeptide helices, the plug was seen to leave the channel and
then, after translocation, return partially to its original location. Also, the pore ring blocked water and
ions before translocation; during translocation, it expanded enough to allow the polypeptide segment through
but did not allow any ions through and only a few water molecules.
Another question perhaps made more complicated by the atomic structure is that regarding
the oligomerization of SecYEβ. While some proteins function only as monomers (single copies), many
form larger structures such as dimers (two copies), tetramers (four copies), or, in general, polymers
(n copies). The SecY/Sec61 complex is known to form dimers and tetramers in nature but it is not
clear why. One original hypothesis was that the channel formed at the center of the four protein complexes.
However, the structure along with other experiments and our simulations seem to demonstrate that the monomer
is capable of forming the channel. The question then remains: what purpose does the tetramer serve? We
have attempted to begin answering this question by simulating a SecYEβ dimer in a back-to-back
conformation (although a front-to-front conformation has also recently been proposed
![]() ).
We discovered that during the simulation, the plug of each SecYEβ became destabilized, much more than in the simulation of a single SecYEβ. Thus, in this way, cooperative
interactions between the two copies of SecYEβ may aid translocation through a single monomer.
).
We discovered that during the simulation, the plug of each SecYEβ became destabilized, much more than in the simulation of a single SecYEβ. Thus, in this way, cooperative
interactions between the two copies of SecYEβ may aid translocation through a single monomer.
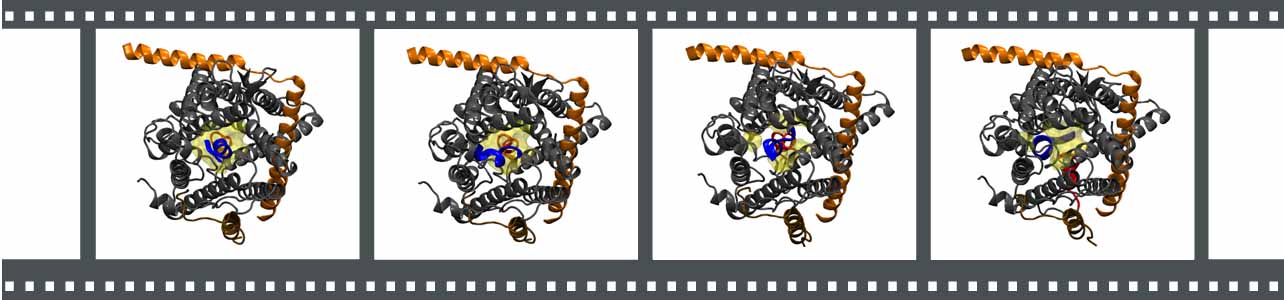
- Click the image above to see a movie made from a simulation of translocation through SecY. Shown from the top, the polypeptide is in blue, the plug in red, and the pore ring residues in yellow. Click here to see the same simulation from the side.
Lateral Gating of the Channel
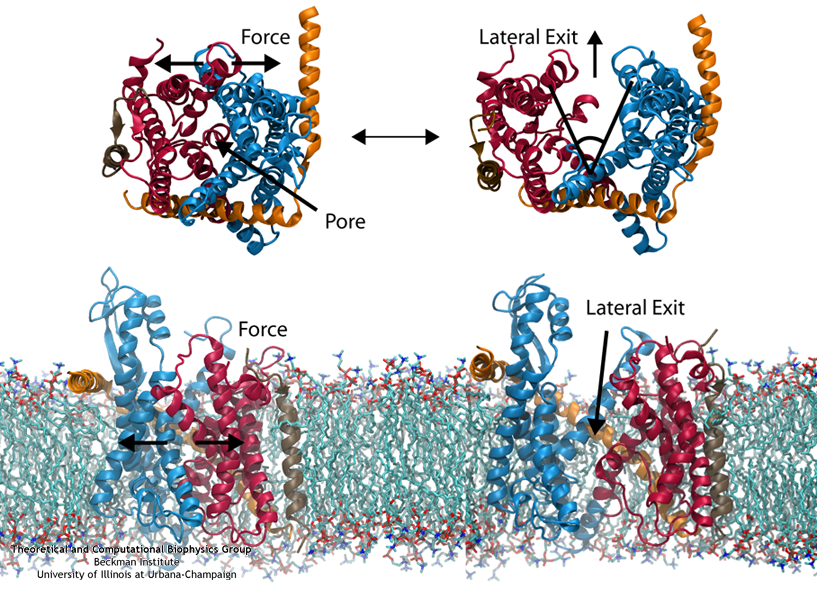
Steered MD was used to force open the lateral gate (click for a larger view).
The other function of the translocon, inserting proteins into the membrane, is accomplished by means of a lateral gate, hypothesized based on the structure
(![]() ).
Simulations of forced gate
opening and relaxation revealed how the structure responds. In particular, the two approximately symmetric halves of SecY were found to stay reasonably stable during opening, agreeing with
the "clamshell-like" behavior proposed previously. Additionally, SecE, the accessory protein surrounding SecY, did not affect the rate of gate closure; this was surprising as SecE had
been assumed to act as a clamp, holding the two halves of SecY together. Also, it was found that by opening the gate, interactions with the plug were broken, freeing it. This is potentially
how gating by a signal sequence occurs, where the inserted sequence pushes (or holds) open the lateral gate, thus freeing the plug and loosening the channel as a whole before translocation.
Click here for a movie of gate opening.
).
Simulations of forced gate
opening and relaxation revealed how the structure responds. In particular, the two approximately symmetric halves of SecY were found to stay reasonably stable during opening, agreeing with
the "clamshell-like" behavior proposed previously. Additionally, SecE, the accessory protein surrounding SecY, did not affect the rate of gate closure; this was surprising as SecE had
been assumed to act as a clamp, holding the two halves of SecY together. Also, it was found that by opening the gate, interactions with the plug were broken, freeing it. This is potentially
how gating by a signal sequence occurs, where the inserted sequence pushes (or holds) open the lateral gate, thus freeing the plug and loosening the channel as a whole before translocation.
Click here for a movie of gate opening.
Also of interest was the behavior of lipids during gate opening. It has been suggested that if the lateral gate opens directly to the bilayer (as opposed to another
translocon next to it
![]() ),
it would become inactivated by lipids flooding the channel. To explore this suggestion in more detail, we simulated SecY with the lateral gate held open to the bilayer, both in an all-atom
representation and a reduced, coarse-grained representation. Surprisingly, we found after up to 1 microsecond, lipids did not invade the channel.
Instead, they clung to hydrophobic gating helices or remained firmly stuck in the bilayer. Given also that a transmembrane segment of an inserting protein probably occupies this region most
of the time, it is probably unlikely then that lipids pose any threat to the channel.
),
it would become inactivated by lipids flooding the channel. To explore this suggestion in more detail, we simulated SecY with the lateral gate held open to the bilayer, both in an all-atom
representation and a reduced, coarse-grained representation. Surprisingly, we found after up to 1 microsecond, lipids did not invade the channel.
Instead, they clung to hydrophobic gating helices or remained firmly stuck in the bilayer. Given also that a transmembrane segment of an inserting protein probably occupies this region most
of the time, it is probably unlikely then that lipids pose any threat to the channel.
Maintaining the Permeability Barrier in the Channel
(left) The system used for simulating the channel. (middle) The channel shown with the native (green) and two mutant (red and blue) plugs. (right) A closeup of the pore ring in one of the mutants with two residues not shown in order to illustrate water permeating the channel (click for a larger version).
While previous simulations indicated that both the pore ring and plug played some role in closing the channel, it remained unclear what their independent roles are. Two
recently solved
structures of mutants further complicated the picture
![]() .
Specifically, two mutants with either half or all of the native plug deleted formed new plugs from the remaining residues. These
mutants demonstrated that having a plug is important for the channel, but how well do the new plugs function? Various experiments indicated they do not function as well as the native plug,
but also do not kill the cell
.
Specifically, two mutants with either half or all of the native plug deleted formed new plugs from the remaining residues. These
mutants demonstrated that having a plug is important for the channel, but how well do the new plugs function? Various experiments indicated they do not function as well as the native plug,
but also do not kill the cell
![]()
![]() .
Electrophysiology experiments demonstrated that the mutant channels permit intermittent permeation of water and ions whereas the native channel is water tight
.
Electrophysiology experiments demonstrated that the mutant channels permit intermittent permeation of water and ions whereas the native channel is water tight
![]() .
However, the crystal structures of the two mutants still show the channel as closed by the pore ring at the channel's center.
.
However, the crystal structures of the two mutants still show the channel as closed by the pore ring at the channel's center.
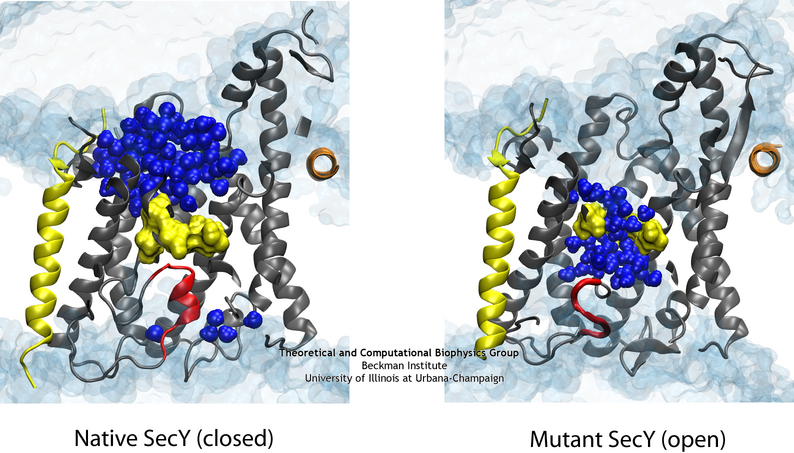
The difference in the permeability is apparent when comparing the native (left) and one of the mutant (right) channels in simulation.
To reconcile the closed structures of the mutants with their experimental permeabilities, we performed multiple equilibrium simulations of the native channel and the two mutants. We measured the permeability of each channel to water, finding increasing diffusive and osmotic permeabilities corresponding to the amount of the plug deleted. The increase in permeability was also found to correspond to an increase in the motions of the new plugs and, in particular, the pore ring residues, suggesting that pore ring flexibility, controlled by the plug, is the primary determinant of channel openness. To confirm this suggestion, we performed a unique computational experiment in which the interactions between water and the plug in the native, closed channel were eliminated, making the plug "transparent" to water. By doing so, we separate the role of the plug as a purely steric barrier to the flow of water from its role as a lock, holding the channel closed. We found in simulations that this hybrid channel permitted almost no water permeation, significantly less than the mutants where even only a fraction of the plug was deleted. Thus it appears that the plug's role as a steric barrier alone is significantly less important than its role holding the pore ring closed.
Binding of a Ribosome to the Channel
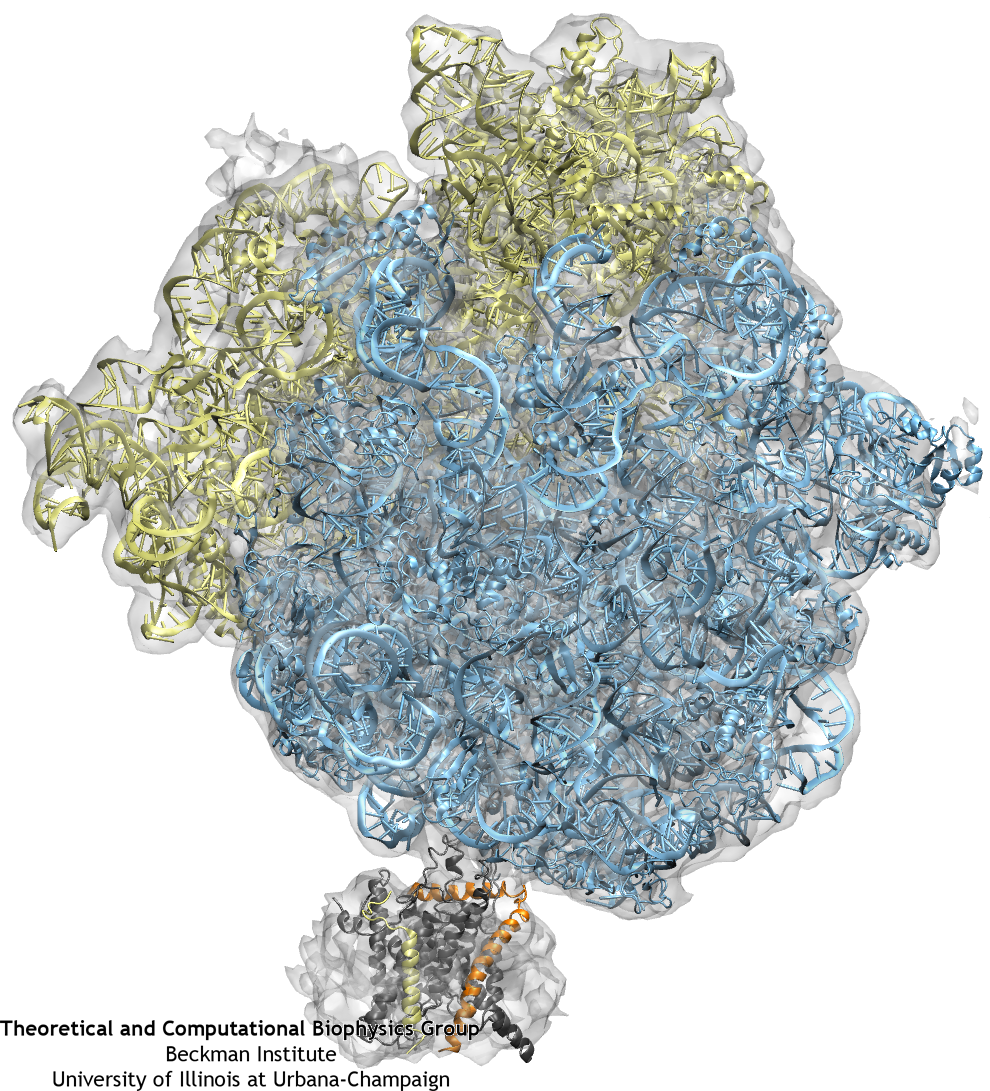
The ribosome (blue and yellow) is docked to the channel (grey, orange, and yellow) as a result of fitting to the cryo-EM map (grey transparent surface).

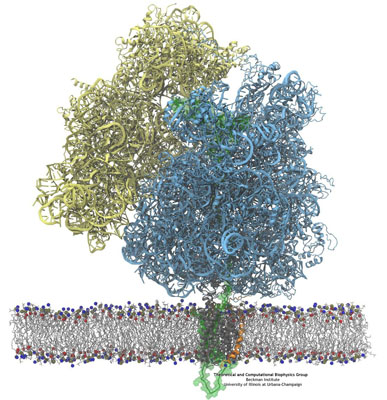
Once the fitting was completed, a system containing ribosome-SecY complex, ions, membrane, and water was built. The system contains 2.7 million atoms in total.
Our previous studies focused on the behavior of the channel alone, under different conditions. However, the channel always works in concert with partners, such as the ribosome, noted above. Nearly all information on ribosome-channel complexes has come from cryo-electron microscopy (cryo-EM) maps, which provides relatively low resolution images of the complex. From these maps, it was deduced that either a dimer or tetramer of SecY/Sec61 binds to the ribosome. Such deductions were based primarily on the size of the density present in the channel region, since specific features were undetectable at their present resolution, leaving open the question of precisely how many copies of the channel are actually present in the complex and in what orientation they are.
Many of the limitations of previous maps have been overcome recently
though. For the first time in 2007, a monomer of SecYEG bound to a ribosome was captured in a
cryo-EM map
(![]() ).
By combining this map with high-resolution crystallographic structures of the separate
ribosome and channel, a high-resolution structure of the complex could be generated. This
combination of data from multiple techniques was accomplished through the use of molecular
dynamics flexible fitting (MDFF), a recently developed method which drives an atomic structure
into a new conformation represented by the cryo-EM map in an MD simulation (more info on our
MDFF webpage). The resulting model of the ribosome-channel
complex revealed, in particular, the sites of ribosome-SecY interaction that maintain the
complex. We found the interactions map to four specific regions of the channel, namely the
two cytoplasmic loops of SecY, the C-terminus of SecY, and the amphipathic helix of SecE (see
the figure on the right). All four sites of interactions are conserved across different
domains of life and even different binding partners. For example, the structure of the
bacterial post-translocation partner, SecA, with SecY also display interactions which involve
the two cytoplasmic loops and C terminus of SecY
(
).
By combining this map with high-resolution crystallographic structures of the separate
ribosome and channel, a high-resolution structure of the complex could be generated. This
combination of data from multiple techniques was accomplished through the use of molecular
dynamics flexible fitting (MDFF), a recently developed method which drives an atomic structure
into a new conformation represented by the cryo-EM map in an MD simulation (more info on our
MDFF webpage). The resulting model of the ribosome-channel
complex revealed, in particular, the sites of ribosome-SecY interaction that maintain the
complex. We found the interactions map to four specific regions of the channel, namely the
two cytoplasmic loops of SecY, the C-terminus of SecY, and the amphipathic helix of SecE (see
the figure on the right). All four sites of interactions are conserved across different
domains of life and even different binding partners. For example, the structure of the
bacterial post-translocation partner, SecA, with SecY also display interactions which involve
the two cytoplasmic loops and C terminus of SecY
(![]() ).
).
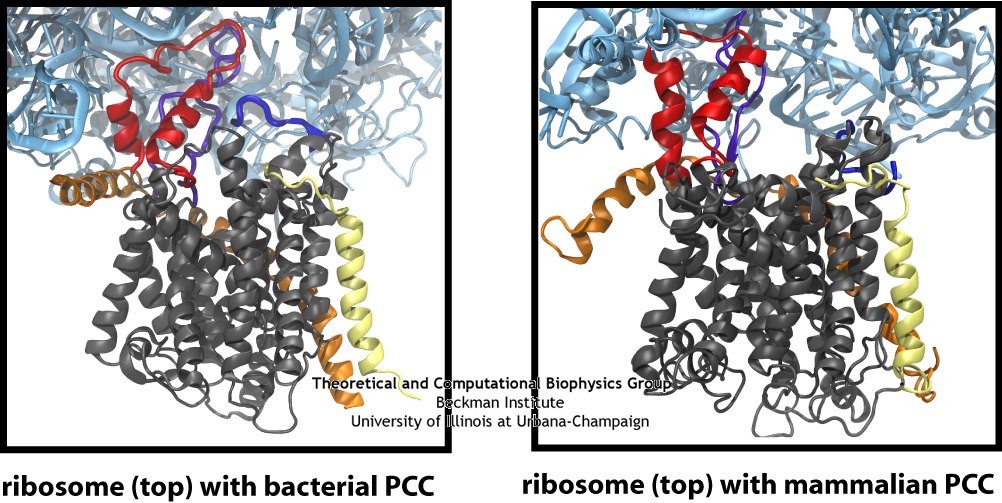
The similarity of the connections between the ribosome and channel in the bacterial and mammalian systems is clearly seen in this side-by-side comparison, particularly the two loops of the channel in red and purple.
Using the atomic-scale structure of a ribosome-channel complex, we prepared a system containing also ions, membrane, and water, requiring altogether 2.7 million atoms. The subsequent simulation, among the largest all-atom simulations every run to date, illustrated the effects of ribosome binding on the channel. We found that the bound ribosome induces a slight destabilization of SecY's plug, possibly representing a "priming" step, preceding insertion of the signal sequence. This effect could be mimicked solely through restraint of the two cytoplasmic loops of SecY with no ribosome present, indicating how multiple channel partners can influence SecY despite all being unique. Additionally, we simulated translocation of a short polypeptide from the ribosome's exit tunnel into the channel. The movement of the polypeptide did not significantly disturb binding between the ribosome and channel, demonstrating that the complex is functional. Finally, we also modeled a back-to-back dimer of the channel bound to the ribosome. The ribosome-SecY-dimer complex was more stable than the ribosome-SecY-monomer complex, suggesting that dimers or tetramers of the channel may form to increase the strength of binding to channel partners, although only one channel is functional.
In addition to the bacterial ribosome-SecY-monomer complex, cryo-EM maps have recently been obtained for eukaryotic ribosome-Sec61-monomer complexes as well
(![]() ,
,
![]() }.
For the highest resolution maps, the detail is sufficient to observe secondary structure elements for the channel itself, something not possible previously. As in the case of the
ribosome-SecY-monomer complex, MDFF was used to fit atomic models of the Sec61 channel to a map of an actively translating mammalian ribosome-Sec61-monomer complex, with a nascent polypeptide
present inside the channel. The fitted structure of the complex revealed connections between ribosome and channel similar to those observed previously for the ribosome-SecY-monomer complex.
Additionally, the N terminus of Sec61α was found to contribute to ribosome binding. This result offers further confirmation that a monomeric channel bound to the ribosome is
functional, and thus places significant limits on any potential role of dimers or tetramers of the channel in translocation.
}.
For the highest resolution maps, the detail is sufficient to observe secondary structure elements for the channel itself, something not possible previously. As in the case of the
ribosome-SecY-monomer complex, MDFF was used to fit atomic models of the Sec61 channel to a map of an actively translating mammalian ribosome-Sec61-monomer complex, with a nascent polypeptide
present inside the channel. The fitted structure of the complex revealed connections between ribosome and channel similar to those observed previously for the ribosome-SecY-monomer complex.
Additionally, the N terminus of Sec61α was found to contribute to ribosome binding. This result offers further confirmation that a monomeric channel bound to the ribosome is
functional, and thus places significant limits on any potential role of dimers or tetramers of the channel in translocation.
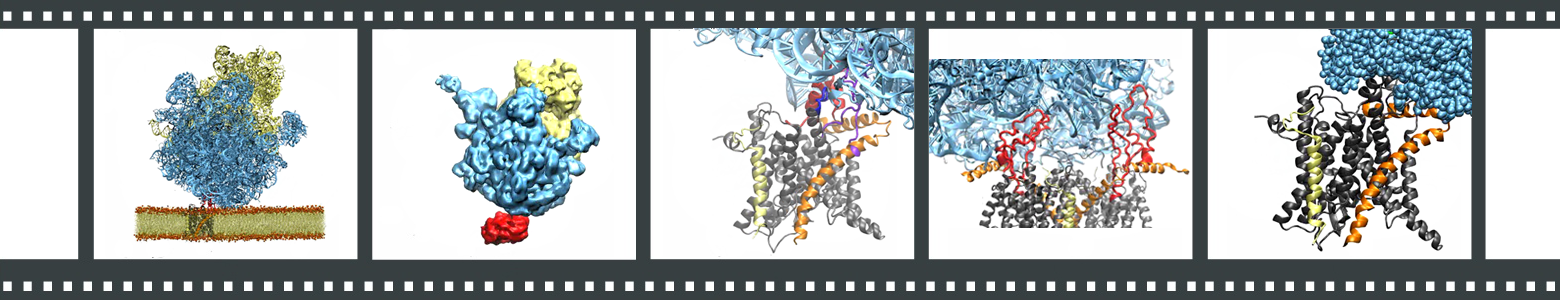
Each frame links to a different movie. (1) Ribosome-SecY-membrane system. (2) Overview of the MDFF procedure. (3) Interactions between ribosome and SecY. (4) Hypothetical ribosome-SecY-dimer complex. (5) Translocation of a small polypeptide from exit tunnel to channel.
Visualizing Membrane Protein Insertion
Prior simulations investigating the membrane-insertion role of the translocon focused on the
channel in the absence of an inserting protein (see "Lateral Gating of the Channel" above).
Evidence of the lateral gate's role in membrane insertion of transmembrane segments has been
largely indirect to date; for example, structures of a SecA-bound SecY
(![]() )
and a Fab-bound SecY
(
)
and a Fab-bound SecY
(![]() )
illustrated a slight opening of the gate, also in the absence of an inserting protein. A
more recent structure of SecY with a symmetry-related copy inserting into the channel also
exhibits modest gate opening
(
)
illustrated a slight opening of the gate, also in the absence of an inserting protein. A
more recent structure of SecY with a symmetry-related copy inserting into the channel also
exhibits modest gate opening
(![]() ).
However, the insertion of a real nascent peptide
remained unresolved.
).
However, the insertion of a real nascent peptide
remained unresolved.
Just as happened previously (see "Binding of a Ribosome to the Channel" above), cryo-electron microscopy was used to capture a novel functional state of the translocon. In the experiments, carried out in the lab of Roland Beckmann at the University of Munich, a ribosome in the act of inserting a nascent protein into the channel was stalled and visualized by cryo EM. The channel was stabilized inside a self-contained lipid membrane, a so-called nanodisc (see our lipoproteins webpage), providing a native environment as opposed to the harsher detergents used to determine prior maps. Using the newly resolved map along with MDFF, we fitted atomic models of the ribosome, translocon, and nascent chain, revealing that the nascent protein's N-terminal signal anchor was lodged in the translocon's fully opened lateral gate, at the border between channel and membrane. The new structure can be downloaded from the PDB database.
The MDFF-derived atomic structure was also used for free MD simulations containing the ribosome-translocon-nascent-chain complex as well as the complete nanodisc. In the simulations, an attraction between the lipids in the nanodisc and the ribosome's helix 59 (H59) developed, with the lipids forming a small ridge. This ridge was found to be an excellent match with some otherwise inexplicable density in the original cryo-EM map, and led to the suggestion that H59 may play a role in aiding the inversion and subsequent insertion of the nascent chain's signal anchor.
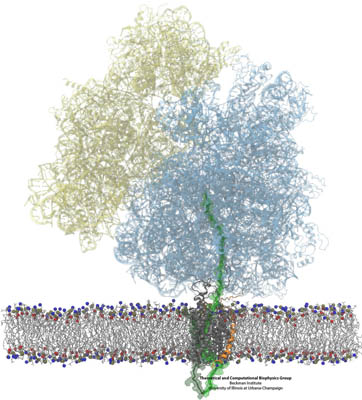
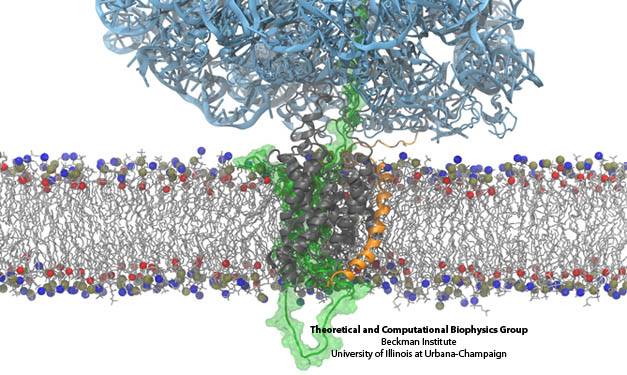
Side-by-side views of the full ribosome-translocon-nascent-chain system. On the right, the path of the nascent chain can be traced from the ribosome's peptidyl transferase center into the channel. The bottom shows a closeup of the channel.
Quantifying Membrane Protein Insertion
 The arginine residue (blue) on a nascent helix (purple) is sampling the grey membrane interior while
still ensconced within the translocon (green, yellow, and orange).
The arginine residue (blue) on a nascent helix (purple) is sampling the grey membrane interior while
still ensconced within the translocon (green, yellow, and orange).
Although we have visualized a transmembrane segment, namely a signal anchor,
in the process of inserting into the membrane, the structure does not address how the segment
decides whether or not to insert. Systematic experiments testing the insertion propensity of various
sequences of amino acids led to the suggestion that insertion is governed by purely by thermodynamics
(![]() ).
An unexpected result from those experiments was that charged residues such as arginine exhibit a
relatively low penalty to be inserted into the membrane. Numerous computational and theoretical studies
suggested a penalty of anywhere from 2 to 7 times what was observed experimentally, leading to confusion
regarding what process was actually being measured.
).
An unexpected result from those experiments was that charged residues such as arginine exhibit a
relatively low penalty to be inserted into the membrane. Numerous computational and theoretical studies
suggested a penalty of anywhere from 2 to 7 times what was observed experimentally, leading to confusion
regarding what process was actually being measured.
One potential source of the discrepancy between the experiments and simulations is in the assumption of the initial reference state. Although both presume that the end result is a transmembrane segment embedded in the bilayer, in the simulations the initial state was often chosen to be in a purely aqueous state, neglecting any potential role of the translocon. We hypothesized that the initial state for membrane insertion is, instead, the interior of the translocon. Using free-energy perturbation simulations, we tested this hypothesis by calculating the energetic cost to move an arginine and a leucine residue from the channel to the membrane. We found that the magnitude of both energies were less than in previous simulations, more closely matching the experimental results. Thus, protein insertion into the membrane likely follows a two-stage process, with the ribosome driving the protein into the channel in the first stage and then it exiting laterally through the gate in the second.
Inserting Simple Proteins into the Cell Membrane
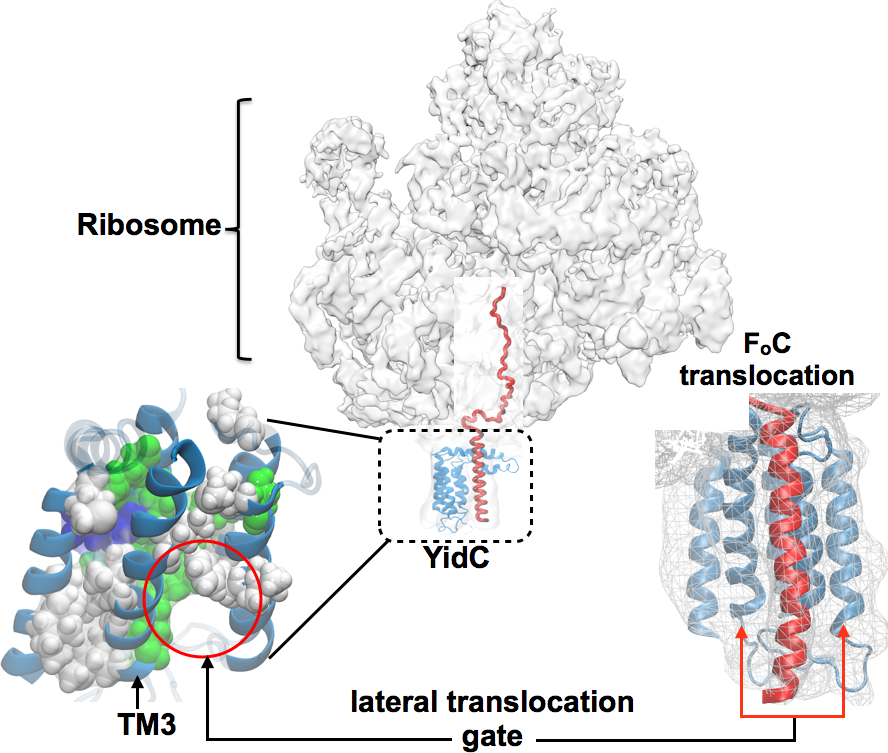
Despite significant advancements in resolving the mechanism of protein insertion via SecY (as enumerated above), structural analysis of the SecY-independent insertion pathway posed severe challenges over the past three decades. The SecY-independent pathway often involves another insertase, YidC, which mediates the insertion of small hydrophobic helices into the membrane, such as the Fo c-ring helices of ATP synthase. Combining cryo-EM, covariance, and biochemical complementarity data with homology modeling and MD simulations one of the first all-atom structures of YidC and its ribosome-bound complex is derived within the membrane environment (here). YidC contains five transmembrane helices one of which, called TM3, is kinked and more loosely bound than the other four (Fig. 6). As the periplasmic side of YidC is tightly packed with aromatic residues from all five helices, the system does not allow protein translocation across the membrane. Rather, the loosely bound and more flexible TM3 together with another helix, TM5, is hypothesized to form a gateway for lateral translocation of the nascent protein chain into the membrane. The quality of this atomic model is validated by its close agreement with a recently published crystal structure of E. Coli YidC (here) and subsequent water-accessibility and mutational studies (here).
|
|
|
|
Lateral translocation of the nascent peptide chain via YidC. The nascent inner membrane proteins synthesized in the ribosome, are co-translationally inserted into the membrane via YidC. A kinked helix (TM3) mediated gate has been hypothesized for lateral protein translocation. The structure of the proposed gate shown at the lower left; polar and hydrophobic residues are shown in green and white respectively. A cartoon of the prospective lateral nascent chain translocation in question is shown at the lower right. |
Publications
Structures
The PDB entry for the cryo-EM-fitted structure of the ribosome-SecY-nascent protein complex is 4V6M.Investigators
Page created and maintained by JC Gumbart.
The contents of this website were reproduced in part with permission from


