Mechanisms of Protein Synthesis by the Ribosome
- Introduction
- Obtaining atomic models of the ribosome at functional states
- How does the ribosome induce the GTPase activity of EF-Tu?
- Nascent-chain mediated translational stalling
- Recognition of the regulatory nascent chain TnaC by the ribosome
- How does the L1 stalk interact with tRNAs in hybrid state?
- Translocation of tRNAs inside the ribosome
- Stalling of the ribosome by SecM
- Mechanism of tetracycline resistance
- An antibiotic action on the ribosome
- Selecting amino acids by their chirality properties
- Stalling of bacterial translation by a nascent protein encoding the antibiotic resistance gene
- Publications
- Investigators
See also the websites on the Dynamics of Protein Translocation and Molecular Dynamics Flexible Fitting (MDFF).
Introduction
The translation of genetic information into proteins is essential for life. At the core of this process lies the ribosome, a quintessential large (2.5-4.5 MDa) molecular machine responsible for translating genetic material into functional proteins. In a growing cell, ribosomes comprise up to half of the net dry weight. Because of its fundamental role in the cell, 50% of all efforts to develop antibiotics target bacterial ribosomes, taking advantage of the structural differences between bacterial and human ribosomes.
The structure and function of the ribosome are fascinatingly complex. Two-thirds of the ribosome consist of ribosomal RNA (rRNA), while over 50 ribosomal proteins make up the rest. The genetic information is delivered to the ribosome by a messenger RNA (mRNA). Transfer RNAs (tRNAs) are adapter molecules, each equipped with an anticodon to match the codons in the mRNA, and charged with an amino acid that corresponds to the anticodon as dictated by the genetic code. The ribosome contains three tRNA-binding sites: A, P, and E (see elongation cycle box, or watch a movie). In addition to mRNA and tRNAs, the ribosome interacts with protein factors such as the elongation factors Tu (EF-Tu) and G (EF-G), that are important players in the so-called elongation cycle. The elongation cycle results in the addition of an amino acid to the nascent peptide chain, and consists of three main steps. In the decoding step, a ternary complex comprised of an aminoacyl-tRNA (aa-tRNA), EF-Tu, and GTP binds to the ribosome, leading to the recognition of the codon by the anticodon. The following step is the peptidyl transfer. Here the peptide chain bound to the P-site tRNA is covalently linked to the amino acid bound to the A-site tRNA. In the translocation step, the position of the mRNA/tRNA complex shifts by one codon, accompanied by a ratchet-like motion of the ribosomal subunits.
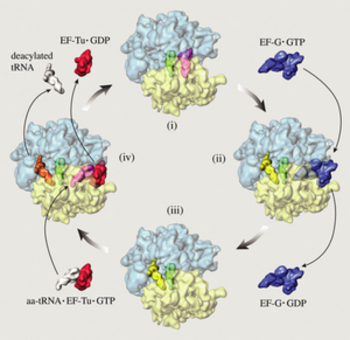 Fig 1. Elongation cycle of protein synthesis. The
ribosome is shown in top view, with the small subunit (transparent
yellow) below the large subunit (transparent blue). (i) The
ribosome in the pre-translocational state with tRNAs in the A
(magenta) and P (green) sites. After spontaneous peptidyl transfer,
the nascent peptide is covalently attached to the A-site tRNA.
(ii) The elongation factor EF-G in complex with GTP (blue) has
bound to the ribosome to facilitate the translocation of tRNAs to
the P (green) and E (yellow) sites. The translocation is induced by
GTP hydrolysis accompanied by large transient conformational
changes in the EF-G and the ribosome. (iii) The release of the EF-G
after GTP hydrolysis leaves the ribosome in the
post-translocational state. It is now ready to accept a new
aminoacyl-tRNA (white) presented to the ribosome by the ternary
complex, comprising, in addition to the new aminoacyl-tRNA, the the
elongation factor EF-Tu and a GTP (red). (iv) The ribosome with a
bound ternary complex (magenta and red) in place. It has been
suggested that when the ternary complex binds to the ribosome, the
E-site tRNA moves further away from the P site to the so-called E2
site (orange). The snapshot shown here is part of the decoding
step, where the aminoacyl-tRNA whose anticodon matches the next
codon in the mRNA is selected to enter the A site, accompanied by
GTP hydrolysis and conformational changes. In case of a match,
EF-Tu with the hydrolyzed GTP and the E-site tRNA leave the
ribosome, leaving it in the pre-translocational state (i). This
figure is adapted from Frank, J. in Conformational Proteomics
of Macromolecular Architechtures. World Scientific
Publishing Comp. Singapore, 2004. Watch a movie of this
process.
Fig 1. Elongation cycle of protein synthesis. The
ribosome is shown in top view, with the small subunit (transparent
yellow) below the large subunit (transparent blue). (i) The
ribosome in the pre-translocational state with tRNAs in the A
(magenta) and P (green) sites. After spontaneous peptidyl transfer,
the nascent peptide is covalently attached to the A-site tRNA.
(ii) The elongation factor EF-G in complex with GTP (blue) has
bound to the ribosome to facilitate the translocation of tRNAs to
the P (green) and E (yellow) sites. The translocation is induced by
GTP hydrolysis accompanied by large transient conformational
changes in the EF-G and the ribosome. (iii) The release of the EF-G
after GTP hydrolysis leaves the ribosome in the
post-translocational state. It is now ready to accept a new
aminoacyl-tRNA (white) presented to the ribosome by the ternary
complex, comprising, in addition to the new aminoacyl-tRNA, the the
elongation factor EF-Tu and a GTP (red). (iv) The ribosome with a
bound ternary complex (magenta and red) in place. It has been
suggested that when the ternary complex binds to the ribosome, the
E-site tRNA moves further away from the P site to the so-called E2
site (orange). The snapshot shown here is part of the decoding
step, where the aminoacyl-tRNA whose anticodon matches the next
codon in the mRNA is selected to enter the A site, accompanied by
GTP hydrolysis and conformational changes. In case of a match,
EF-Tu with the hydrolyzed GTP and the E-site tRNA leave the
ribosome, leaving it in the pre-translocational state (i). This
figure is adapted from Frank, J. in Conformational Proteomics
of Macromolecular Architechtures. World Scientific
Publishing Comp. Singapore, 2004. Watch a movie of this
process.
|
Due to great advances in the structural resolution of the ribosome, an impressive feat given its large size, the system is considered one of the hottest focal areas in molecular cell biology today. During the process of translation, the ribosome undergoes several conformational changes and binds to different factors that catalyze specific reactions. As detailed below, techniques to determine structure of the ribosome can only image snapshots of the ribosome, often at medium to low resolution. Atomic details of the interactions between the factors and the ribosome, along with a dynamic description of the conformational changes of the ribosome itself, are crucial to understanding its function.
Our goal is to provide a complete description of the structural dynamics of translation using computational biology. Eventually, we would like to be able to show you an animation much like this beautiful illustrative movie but with every frame based on physically realistic simulations that incorporate experimental data.
Obtaining atomic models of the ribosome at functional states
|
|
|
|
Fig. 2. Detail of a fitted atomic model into the EM density using MDFF. |

Due to its sheer size and complexity, the ribosome presents an outstanding challenge for traditional methods for high-resolution structure determination such as X-ray crystallography and nuclear magnetic resonance spectroscopy. X-ray crystallographers have conquered this challenge: today, the Protein Data Bank has several structures of entire ribosomes from different laboratories. However, these structures remain difficult to obtain for factor-bound ribosomes, which are key to understand the dynamics of translation.
Cryo-electron microscopy (cryo-EM) provides an alternative to obtain structural information of the ribosome in many different functional states. Cryo-EM produces three-dimensional density maps of ribosomes at medium resolution (7-12Å) without the need to purify large amounts of sample or to form highly ordered crystals, both required in X-ray crystallography. This technique thus can capture the ribosome at different conformational states, as shown in Fig. 1. However, these maps don't reach atomic resolution, needed to understand the function of the ribosome in detail along its functional cycle.
In order to reach atomic resolution, we have developed a technique called molecular dynamics flexible fitting (MDFF) to morph atomic-resolution structures emerging from X-ray crystallography into cryo-EM maps. Details of the MDFF method can be found here. Fig. 2 shows a detail of an atomic model flexibly fitted into a cryo-EM map of the ribosome.
We have teamed with the laboratories of Joachim Frank and Roland Beckmann, experts on ribosome structure determination by cryo-EM, to study the dynamics of the ribosome.
How does the ribosome induce the GTPase activity of EF-Tu?
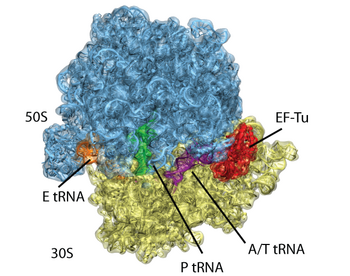
|
|
|
Fig. 3. Atomic model of the ribosome bound to factors derived from a 6.7-Å EM map. The EM data is shown in transparent surface, with atomic model generated with MDFF showed in cartoon. |
During elongation, the mRNA-programmed ribosome is presented with an amino acid by a tRNA in complex with elongation factor Tu (EF-Tu) and GTP. The tRNA assumes the bent A/T position, storing elastic energy. If the ribosome recognizes the right codon-anticodon interaction, GTPase activity in EF-Tu is stimulated over 75Å away. How the ribosome induces GTP hydrolysis has been subject of intensive study. We now know that GTP hydrolysis takes place through an in-line attack on the gamma phosphate by a water molecule, and that His84 is the catalytic residue.
|
|
|
|
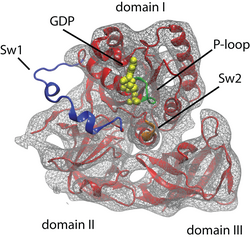
Fig. 4. Atomic model of a ribosome-bound EF-Tu. The switch regions are highlighted: Switch I (Sw1) in blue, Switch II (Sw2) in orange, and P-loop in green. |
In order to understand GTP hydrolysis in EF-Tu, we studied the ribosome-induced EF-Tu conformational changes that trigger GTP hydrolysis on the factor as revealed by cryo-electron microscopy (cryo-EM) studies. We applied MDFF to obtain an atomic model of a 6.7-Å cryo-EM map of the pre-accommodated E. coli 70S ribosome bound to the Phe-tRNAPhe:EF-Tu:GDP ternary complex stalled by the antibiotic kirromycin (kir), showed in Fig. 3.
EF-Tu has three important regions that play a prominent role in its GTPase activity: Switch I (EF-Tu residues 40--62; E. coli numbering) Switch II (80--100) and P-loop (18--23). All of these regions undergo characteristic conformational changes in EF-Tu. Crystal structures of EF-Tu in the GTP form display a "hydrophobic gate" formed by residues Val20 (in the P-loop) and Ile60 (In Switch I) that controls access to the GTP binding pocket. A schematic representation of this gate is shown in Fig 5A, the crystal structure shown in Fig. 5B. His84 (in Switch II) needs to enter through this gate to perform its catalytic role. Thus, this gate must be opened when the right codon-anticodon interaction is recognized by the ribosome. A crystal structure of EF-Tu outside the ribosome, but bound to the antibiotic aurodox that is thought to simulate interaction with the ribosome, displays an open gate (Fig. 5D). This antibiotic binds to EF-Tu and stimulates GTPase activity, preventing EF-Tu from binding the ribosome, and therefore preventing translation and killing the cell. The fact that an open gate is found in the case of enhanced GTPase activity hints that this mechanism is used by the ribosome as well.
|
|
|
|
Fig. 5. Hydrophobic gate of EF-Tu. (A) Schematic representation of the gate. (B) The closed gate revealed by an X-ray structure of EF-Tu free from the ribosome (PDB 1OB2). (C) The open gate revealed by MDFF (PDB 3FIH). (D) The open gate present in an antibiotic-bound X-ray structure (PDB 1HA3). |
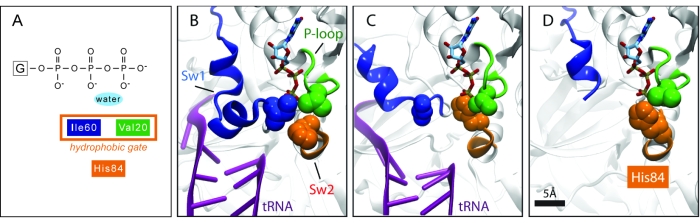
But how does the ribosome open this hydrophobic gate? Let's look at each wing of the gate:
Interaction between the P-loop and SRL
|
|
|
|
Fig. 6. Interaction between the P-loop (green) and the SRL in the 50S (blue). |
Interaction between Switch I and the 16S rRNA
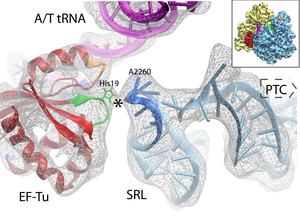
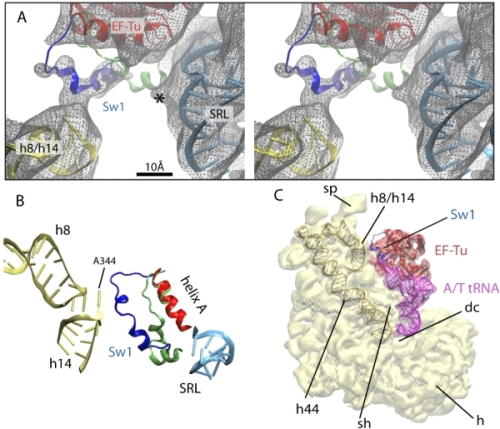
The other wing of the hydrophobic gate (Ile60) is part of Switch I, which due to its flexibility sometimes is difficult to observe in structural studies. Our data and analysis reveal that Switch I interacts with the junction formed by helices h8 and h14 of the 16S rRNA (Fig. 7A,B). This interaction involves an opening of the hydrophobic gate, as shown in Fig. 5C. The contact between Switch I and the 16S rRNA h8/h14 junction could serve as a pathway for signal transduction to/from the decoding center through helix h44 of the 16S rRNA; this helix interacts with the decoding center at one end and with the h8/h14 junction at the other (Fig. 7C).
|
|
|
|
Fig. 7. Interaction between the Switch I of EF-Tu and the 16S rRNA. (A) Stereo view of the different conformations of Switch I when bound (blue) and unbound (lime) to the ribosome. The EM map is shown in gray mesh. (B) Detail of the interaction. Elements of the ribosome-bound EF-Tu (red and blue) and the 30S (yellow) and 50S (cyan) subunits are shown along with the crystal structure of the Switch I in the ribosome-unbound EF-Tu (lime; PDB 1OB2). (C) Overview of the ternary complex (EF-Tu in red, tRNA in purple) bound to the 30S subunit (yellow). |
Open Sesame
Once the gate is opened, the side chain of His84 can reach the GTP binding pocket and activate hydrolysis, as shown in Fig. 5C; note the similarity to the conformation seen in the aurodox-bound structure of EF-Tu outside of the ribosome, shown in Fig. 5D.EM Map and Atomic Models
The atomic coordinates have been deposited in the Protein Data Bank, with PDB ID codes 3FIH (30S and factors) and 3FIK (50S). The cryo-EM density map has been deposited to the 3D-EM database , with code EMD-5036.Get all the files and a VMD saved state to visualize them together here (107 Mb).
Nascent-chain mediated translational stalling
Certain nascent peptide chains are able to regulate ribosome function while they are still being synthesized, i.e., when they are still inside the ribosomal exit tunnel. One of the classical examples is TnaC, a leader peptide of the tryptophanase operon in E. coli. At high concentrations of tryptophan, TnaC stalls the ribosome, inhibiting termination of its synthesis. Through an intricate gene regulatory mechanism, stalling ultimately leads to the expression of genes responsible for degrading tryptophan.
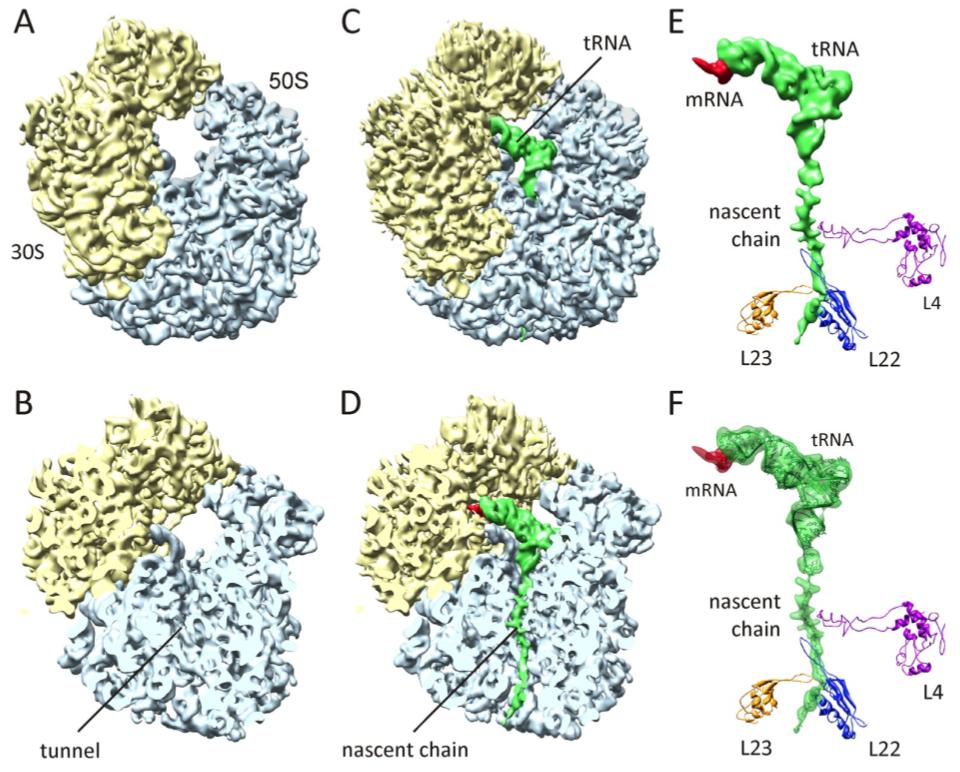
The structural basis for TnaC-mediated translational stalling was addressed by obtaining a 5.8-Å cryo-EM map of the ribosome stalled by TnaC and high concentrations of tryptophan (Fig. 8). The cryo-EM data shows that the nascent chain adopts a distinct conformation in the exit tunnel. We applied MDFF to obtain an atomic model of the entire ribosome and the stalling nascent chain (Fig. 8F). The model allowed us to map the contacts between TnaC and the exit tunnel, as well as propose possible communication pathways that would lead to inactivation of the catalytic center of the ribosome (the so-called peptidyltransferase center, or PTC). One of the main findings was that two critical ribosomal residues at the PTC adopt conformations that are incompatible with cohabitation by release factors, which catalyze termination of protein synthesis.
|
|
|
|
Fig. 8. Cryo-EM reconstruction of the TnaC:70S complex. (A and B) Cryo-EM reconstruction of the control E. coli 70S ribosome at 6.6-Å resolution. (C and D) The 5.8-Å density of the 70S:TnaC complex. (E) Isolated density for mRNA, tRNA, and nascent chain, with the positions of selected ribosomal proteins shown. (F) Same as (E), but with the atomic model obtained using MDFF shown within the transparent density. |
Recognition of the regulatory nascent chain TnaC by the ribosome
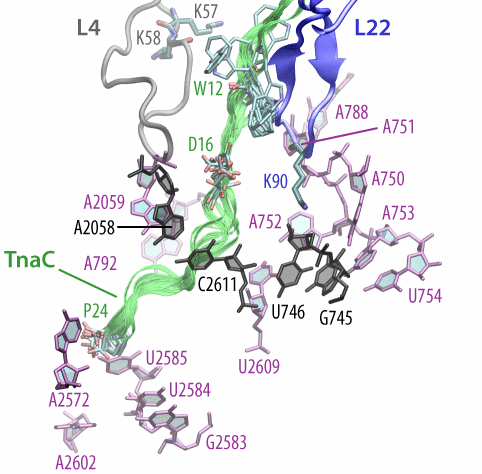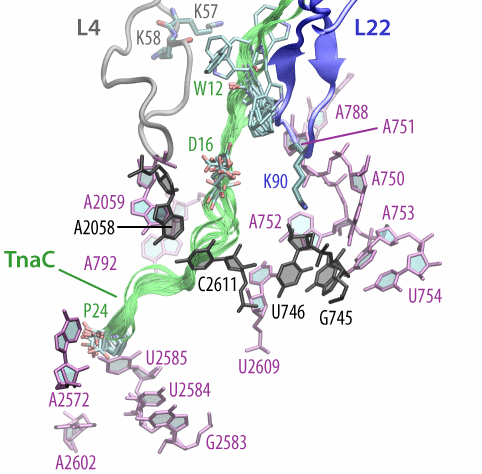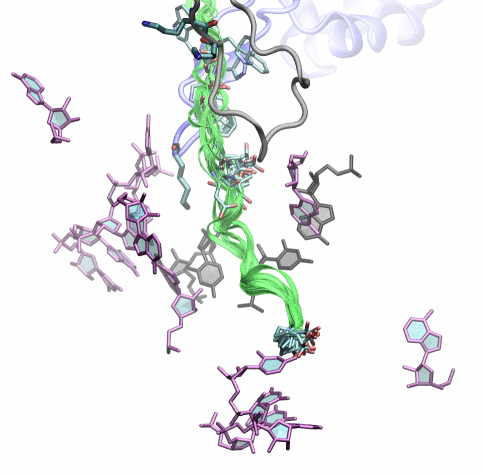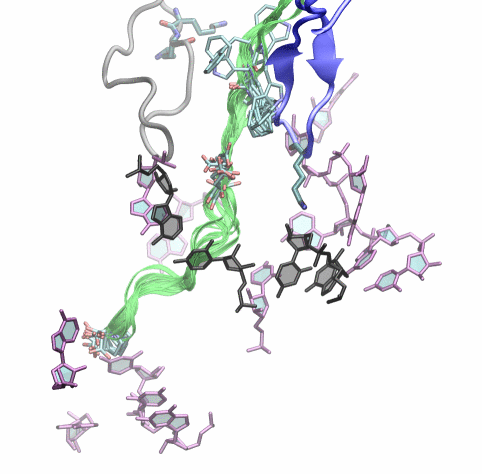
The atomic model of the ribosome stalled by TnaC, presented above, revealed the mechanism of translational arrest. Due to the limited resolution, however, the TnaC side chains and their interactions with the ribosome could not be unambiguously visualized. In fact, 10 possible model of the TnaC nascent chain inside the ribosome were obtained (Fig. 9, left). In order to characterize the precise interactions that allow the ribosome to specifically recognize the TnaC sequence, we performed extensive molecular dynamics (MD) simulations of each of the 10 different models of TnaC inside the exit tunnel for a total aggregate time of over 2 microseconds.
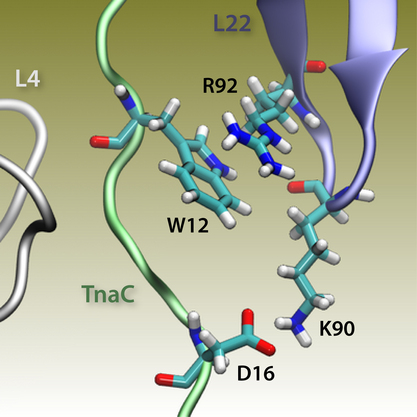
The simulations allowed us to characterize the precise interactions that allow the ribosome to recognize the TnaC sequence inside the exit tunnel (Fig. 9, right). The critical TnaC residue Trp12 is recognized via a cation-pi interaction by Arg92 of ribosomal protein L22. Since classical MD force fields fail to accurately describe such interactions, the study was complemented by quantum chemistry calculations. The MD simulations also revealed that Asp16, another critical TnaC residue, is recognized by Lys90 of L22, also essential for stalling. Moreover, bioinformatic analyses of naturally occurring proteins that share the TnaC consensus sequence suggested mutations that may alleviate TnaC-mediated stalling.
|
|
|
|
Fig. 9. (Left) Structure of TnaC inside the exit tunnel. Shown are 10 different TnaC models with critical residues highlighted. Also shown are ribosomal residues with available biochemical data regarding TnaC-mediated stalling. Click the image to see a movie of a rotating structure (size: 22 M). (Right) Recognition of critical TnaC residues by the ribosome reveal by extensive molecular dynamics simulations. Shown is a cation-pi interaction between critical residue Trp12 (TnaC) and Arg92 (ribosomal protein L22), as well as a salt bridge between critical residues Asp16 (TnaC) and Lys90 (L22). |
|
How does the L1 stalk interact with tRNAs in hybrid state?
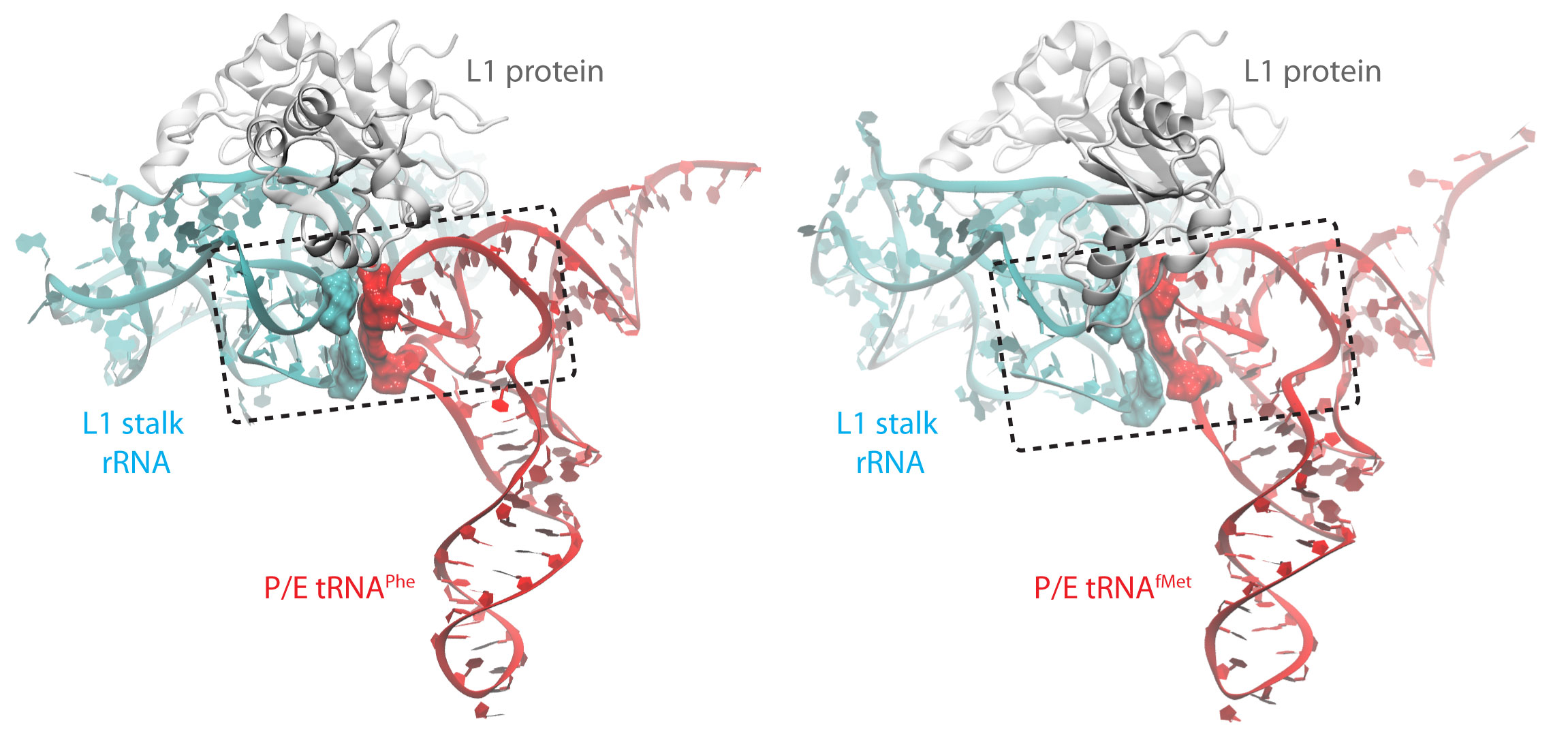
After the accommodation of the tRNA brought by EF-Tu and successful peptidyl transfer, the ribosome is able to spontaneously undergo an intersubunit rotation (Fig. 1 (i) to (ii)). In this state the tRNAs assume a hybrid occupation relative to the ribosome: while the anticodon is still located in the original site on the small subunit, the other end of the tRNA is shifted to the next binding site on the large subunit generating so called A/P and P/E hybrid states. The P/E tRNA engages in interactions with the L1 stalk, a very dynamic part of the ribosome. These interactions are thought to stabilize the hybrid state. Interestingly, based on FRET experiments it seems that the initiator tRNA interacts weaker with the L1 stalk.
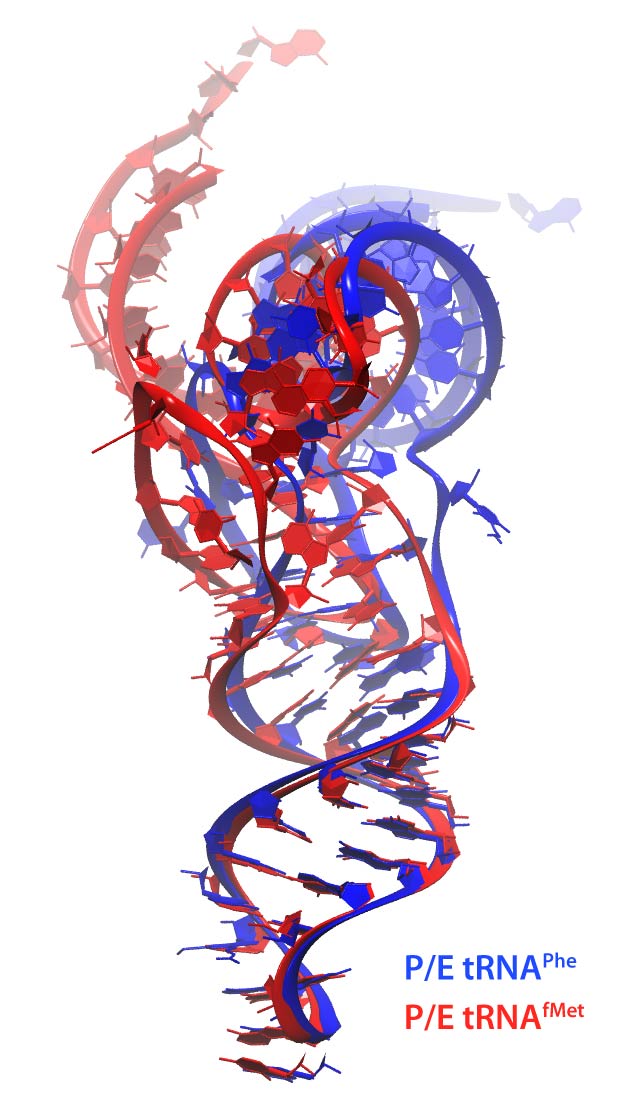
Using MDFF and equilibrium MD we addressed the nature of the interactions between the L1 stalk and a tRNA in the hybrid P/E state. In particular, we compared the behavior of the initiator tRNAfMet versus the elongator tRNAPhe. Not only do the two tRNAs assume different conformations within the ribosome (Fig. 10), they interact differently with the L1 stalk. While for both tRNAs a peculiar stacking with the ribosomal RNA of the L1 stalk is observed, it is less pronounced for the initiator tRNA compared to an elongator tRNA (Fig. 10). Interestingly, the behavior of the tRNAs is strongly impacted by their respective modification patterns.
|
|
|
|
Fig. 10. (Left) Superimposed structures of tRNAPhe and tRNAfMet within the ribosome. (Center) The interactions between tRNAPhe and the L1 stalk. The stacking interactions are shown as surfaces within the dotted box. (Right) The corresponding interactions for tRNAfMet. |
|
Translocation of tRNAs inside the ribosome
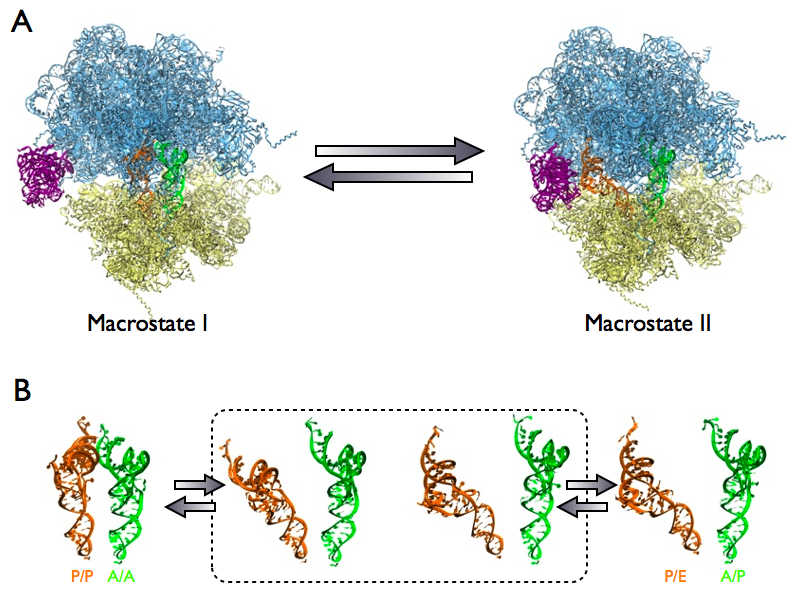
During the spontaneous intersubunit rotation, or the so-called ratcheting, of the ribosome, tRNAs inside the ribosome adopt two different conformations, the classical (A/A and P/P) state and the hybrid (A/P and P/E) state. Together with the motion of L1 stalk, these conformations are collectively termed as two distinct states, namely "macrostate I" (unratcheted ribosome, classical tRNAs and open L1 stalk) and "macrostate II" (ratcheted ribosome, hybrid tRNAs and a closed L1 stalk), as shown in Fig 11A. The transition from "macrostate I" to "macrostate II" is essential for translocating the tRNAs inside the ribosome such that the site for tRNA bearing the next amino acid can be vacated. The existence of intermediate states in between these two conformations is of great debate due to lack of structural data.
Application of MDFF to cryo-EM data of the ribosome revealed two previously unseen intermediate states (Fig 11B). The identification of these new intermediate states help elucidating the pathway of transition, in particular the formation of hybrid tRNAs inside the ribosome. Our results also support the idea that the ribosome is employing a Brownian mechanism to progress through the distinct states during the translocation of mRNA-tRNAs.
|
|
|
|
Fig. 11. (A) Two distinct states of ribosomal complex during translocation of mRNA-tRNA. Macrostate I has an unratcheted ribosome, classical tRNAs (orange and green) and an open L1 stalk (purple). Macrostate II has a ratcheted ribosome, hybrid tRNAs and closed L1 stalk. (B) Two new intermediates (in dashed-lined box) identified by cryo-EM analysis and MDFF. |
Stalling of the ribosome by SecM
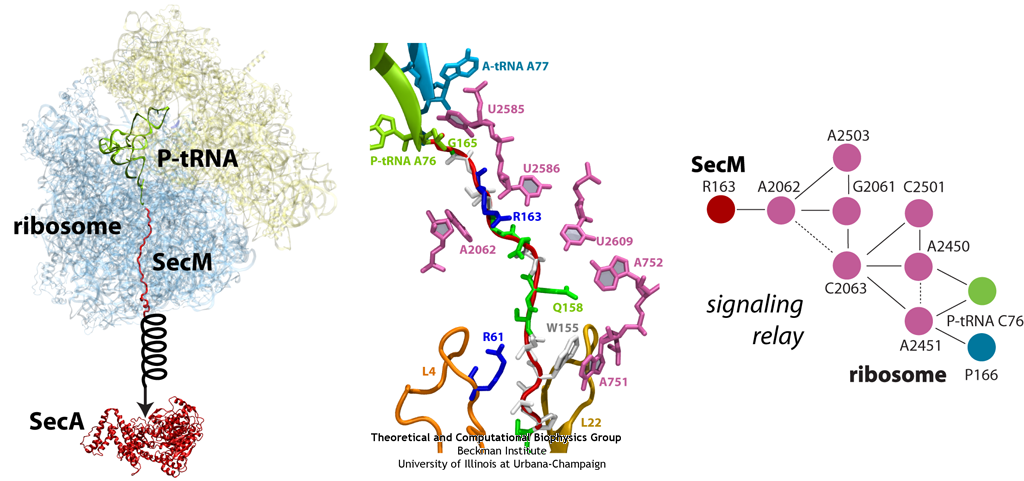
Similar to TnaC described above, the peptide SecM exists solely to stall the ribosome synthesizing it. But unlike TnaC, which also requires the presence of high levels of trytophan, SecM has an intrinsic stalling capability. Stalling of the ribosome synthesizing SecM provides time for a downstream RNA helix on the same mRNA strand to unwind. Unwinding of this helix then allows for a new ribosome to bind and synthesize a new protein, SecA, a bacterial ATP-driven translocase that aids the passage of nascent proteins across membranes in conjunction with SecY (see also Dynamics of Protein Translocation). When sufficient levels of SecA have been reached, SecA interacts with the SecM-stalled ribosome to pull on SecM, freeing it and allowing translation to resume (illustrated schematically in Fig. 13). SecM, which serves no other purpose than to stall the ribosome, is released into the cell and degraded.
Much of what is known about SecM stalling comes from biochemical experiments, in which every amino acid in SecM's sequence has been mutated and the resulting effects measured. These experiments revealed few residues as critical, although one stands out in every species as invariable: arginine 163 (R163, see Fig. 13 below). However, until recently, little was known about the precise mechanisms at work. A cryo-EM map of a SecM-stalled ribosome revealed a shifted linkage in the PTC between the P-site tRNA and the SecM peptide. Although the shift was only 0.2 nm, it was hypothesized to be sufficient to inhibit peptide-bond formation, preventing synthesis of the remainder of SecM.
In order to elucidate precisely how SecM stalls the ribosome, we first applied MDFF to the cryo-EM map of the SecM-stalled ribosome to develop an atomic model of the complex. Next, we carried out extensive simulations of this wild-type model along with mutants of both the ribosome and SecM known to disrupt stalling. We discovered a putative communication pathway between the critical R163 and the PTC that causes the observed shifted linkage; the pathway is shown schematically in Fig. 13. Finally, through steered MD simulations in which we pulled on the N-terminal end of SecM outside the ribosome, we determined that the pulling action of SecA can break the crucial interactions identified in our previous simulations and, thus, alleviate stalling.
|
|
|
|
Fig. 13. (left) Structure of the SecM-stalled ribosome determined with MDFF. SecA is shown for visualization purposes only. (middle) Atomic-scale view of SecM in the ribosome's exit tunnel, near the PTC at the top. Residues involved in ribosome-SecM interactions are labeled. (right) Schematic of the communication pathway connecting SecM to the ribosome's PTC. |
An antibiotic action on the ribosome
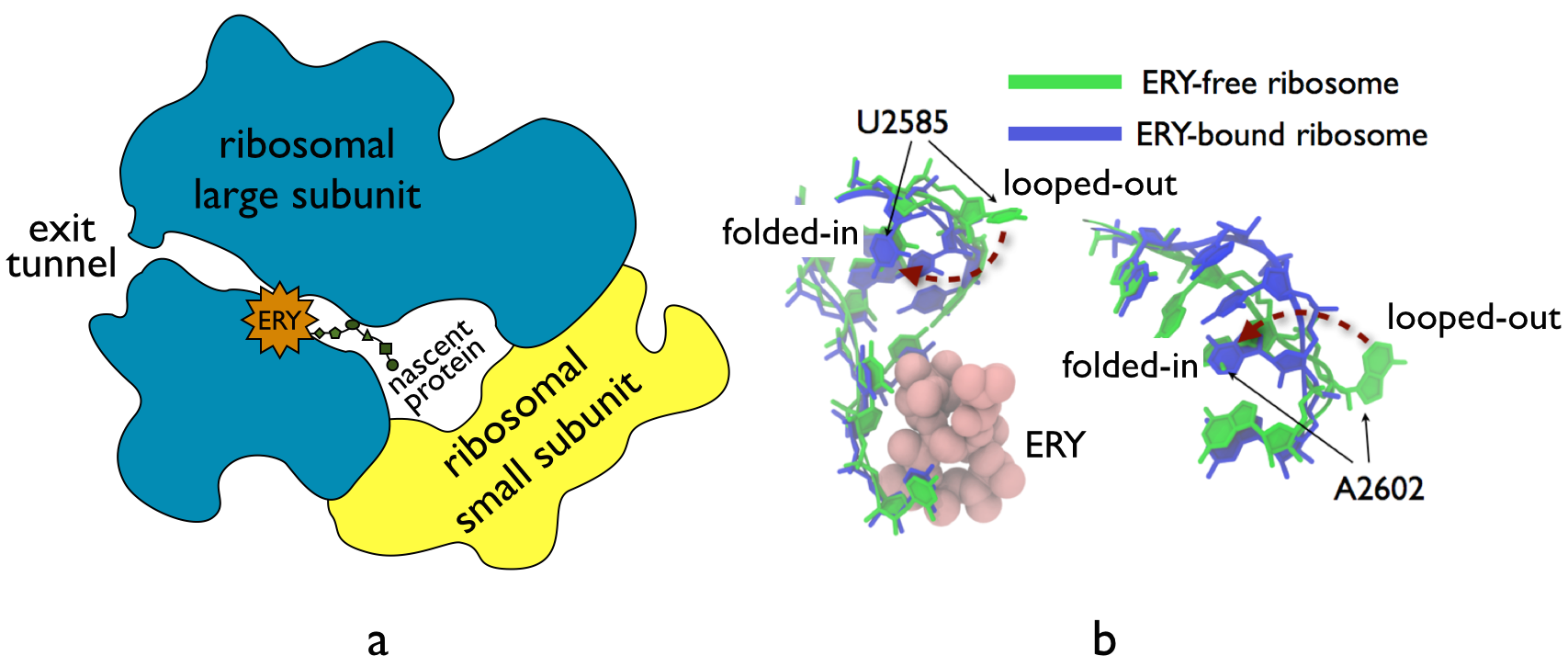
The bacterial ribosome is the target of over 50% antibiotic drugs, for example, the clinically important macrolide family, including the widely-prescribed erythromycin (ERY) which is on the WHO essential medicines list. The antibiotic action of macrolide drugs has been known for over 50 years, however, the molecular mechanisms underlying the effects of these drugs are still unknown. The increasingly serious problem of bacterial resistance against antibiotic drugs is developing now into a major global concern, because no new antibiotic drugs have been developed for nearly 30 years while new strains of bacteria have evolved to be resistant to existing drugs. To promote novel designs of the next generation of antibiotics that are more effective and less resistance-inductive, we investigated the molecular mechanisms underlying the antibiotic action of macrolide ERY on bacterial ribosomes. Our results made possible by large-scale molecular dynamics simulations on the BlueWaters supercomputer showed that the ERY drug takes antibiotic effects by altering the structure of the bacterial ribosome.
The antibiotic drug ERY acts as a protein synthesis inhibitor. As shown in Fig. 14a, ERY binds to the ribosomal exit tunnel of bacterial ribosomes. It was previously believed that the presence of the nascent protein, connecting the drug binding site and the core of the ribosome, was necessary for the action of macrolide drugs. However, we found that the macrolide drugs may act on the ribosome directly without the presence of the nascent protein. First, our collaborators in the Mankin lab at the University of Illinois at Chicago performed SHAPE biochemical modification experiments with both ERY-bound and drug-free empty ribosomes, i.e., ones without the nascent protein, we found that the ribosomal nucleotide U2585 changes conformations after drug binding. We then modeled accordingly an ERY-bound empty ribosome and a drug-free empty ribosome based on the complete crystal structures of ribosomal complexes (PDB codes: 3OFO/3OFR and 2AVY/2AW4, respectively). We found that the antibiotic drug ERY reproducibly induced conformational changes of the universally-conserved ribosomal nucleotides U2585 and A2602 (Fig. 14b), in agreement with the experiments. In particular, flipping of U2585 and A2602 from a looped-out orientation, required for aligning tRNA substrates to prepare the peptide-bond transfer, to a folded-in orientation was observed in the ERY-bound ribosome simulations. We note that A2602 in the looped-out orientation is also required to prevent premature nascent protein release. By contrast, the two nucleotides predominantly assumed looped-out orientation in drug-free ribosome simulations.
In summary, our MD simulations show that the macrolide ERY may trigger antibiotic effects to the bacterial ribosome through allosterically altering the ribosome structures, rather than through interplay with the nascent protein as previously suggested. This finding unveils a principally new view of the antibiotic action of macrolides to the bacterial ribosomes.
|
|
|
|
Fig. 14. a. The ribosome is composed of a large subunit and a small subunit. The nascent protein elongates from the core of the ribosome and egresses through the ribosomal exit tunnel to the outside of the ribosome. The macrolide ERY binds to the exit tunnel. b. molecular dynamics simulations show drug-induced nucleotide-flipping in the ribosome. |
Selecting amino acids by their chirality properties
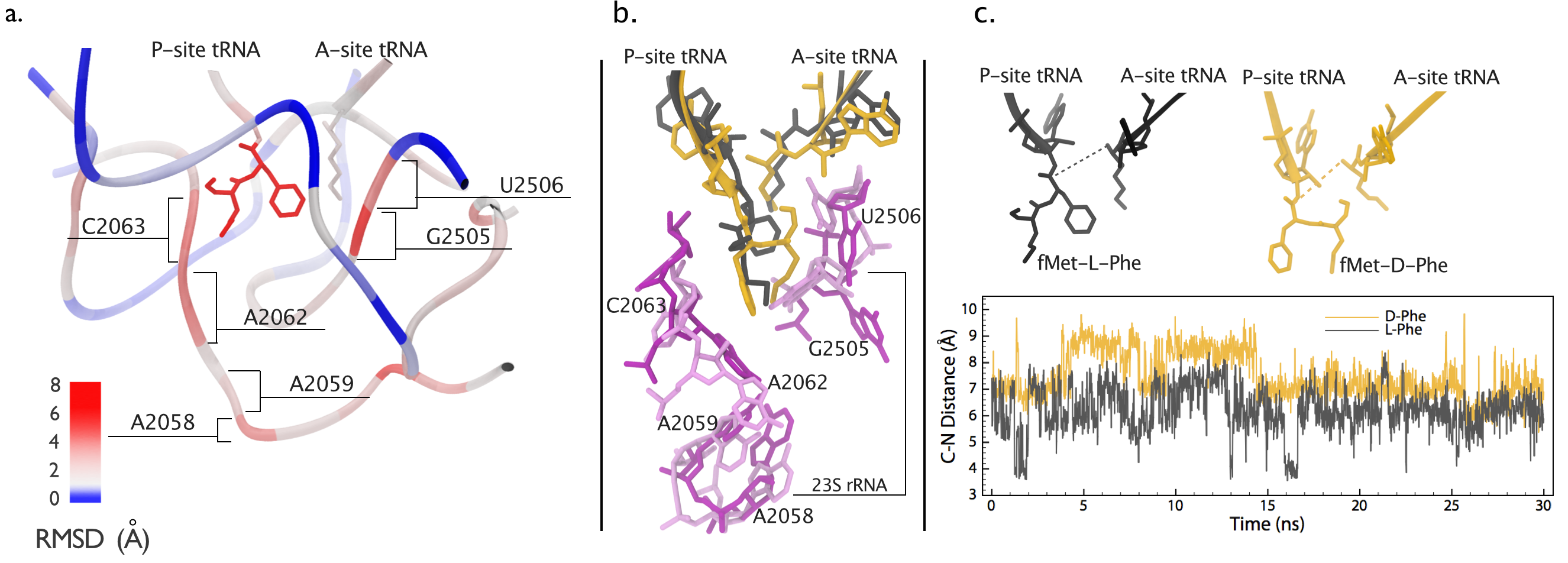
The ability of the ribosome to synthesize proteins during translation using twenty plus chemically diverse amino acids is attributed to the presumed silent role of the amino acid in an aminoacyl-transfer RNA (aa-tRNA) selection process that is governed by messenger RNA (mRNA) codon-tRNA anticodon interactions. Aminoacyl-tRNA synthetases (aaRSs), however, are capable of misacylating tRNAs with D-amino acids which may share the same codon-anticodon pairs. Indeed, several D-amino acids, including D-Ser in the human central nervous system, are present at very high concentrations in vivo. Nevertheless, protein synthesis occurs exclusively with achiral- or L-amino acids, thus reflecting a need for regulatory mechanisms that prevent misincorporation of D-amino acids during translation to guarantee the fidelity of translation.
The need for such regulatory mechanisms leads to a hypothesis that the chirality of the amino acid may be a determinant of the fidelity of aa-tRNA selection. To assess the role of amino acid chirality in the fidelity of protein synthesis, our experimental collaborators performed biochemical assay experiments and found that the ribosome indeed is able to incorporate D-amino acid. However, after the addition of a D-amino acid to the nascent protein the translation is stalled. In order to reveal the molecular mechanism underlying translational stalling by the incorporation of D-amino acid, we performed all-atom simulations. Our computational results shows that the incorporated D-amino acid disrupts the structure of ribosomal peptidyl-transferase center (PTC). The rearrangement of the PTC structure results in an environment that is not optimal for peptide-bond formation. Further chemical protection experiments also confirms a large conformational change inside the PTC after a D-amino acid is incorporated to the nascent protein, as shown in the Fig. 15 below. ribosome readily discriminates between L- and D-amino acids within its peptidyl-transferase center (PTC) during a heretofore undetected step in the mechanism of peptide-bond formation. Further chemical protection experiments confirmed These findings have important implications for the incorporation of unnatural amino acids into polypeptides using misacylated tRNAs, for our understanding of the mechanism of protein synthesis, and for the unraveling of growing links between tRNA misacylation and human neurodegenerative diseases
Finally, our joint efforts with experimentalists have discovered a heretofore undetected step in the mechanism of ribosomal translation which is responsible for selecting L-amino acids instead of D-amino acids. For research details, please refer to this paper, which is recently selected as an F1000Prime paper.
|
|
|
|
Fig. 15. a. structure comparison of the peptidyl-transferase center (PTC) containing either an L- or a D-Phe amino acid in the ribosome. The structure is colored by per-residue values of Root Mean Squared Displacement between the two systems. b. nucleotides with large structural differences are shown. Peptides containing an L-amino acid and their carrier tRNA is colored in black. Peptides containing a D-amino acid and their carrier tRNA is colored in gold. PTC nucleotides are coloered in dark purple and light purple for L- and D-amino acid systems, respectively. c. The changes of distances between the electrophilic carbonyl carbon in the P site and the nucleophilic nitrogen of the amino group in the A site (C-N distance) are shown for L- and D-amino acid systems. The color scheme is the same as b. |
Stalling of bacterial translation by a nascent protein encoding the an tibiotic resistance gene
|
|
|
|
Fig. 16. MDFF-derived all-atom model of an ERY-bound ribosome with the ermb nascent polypeptide in the tunnel. |
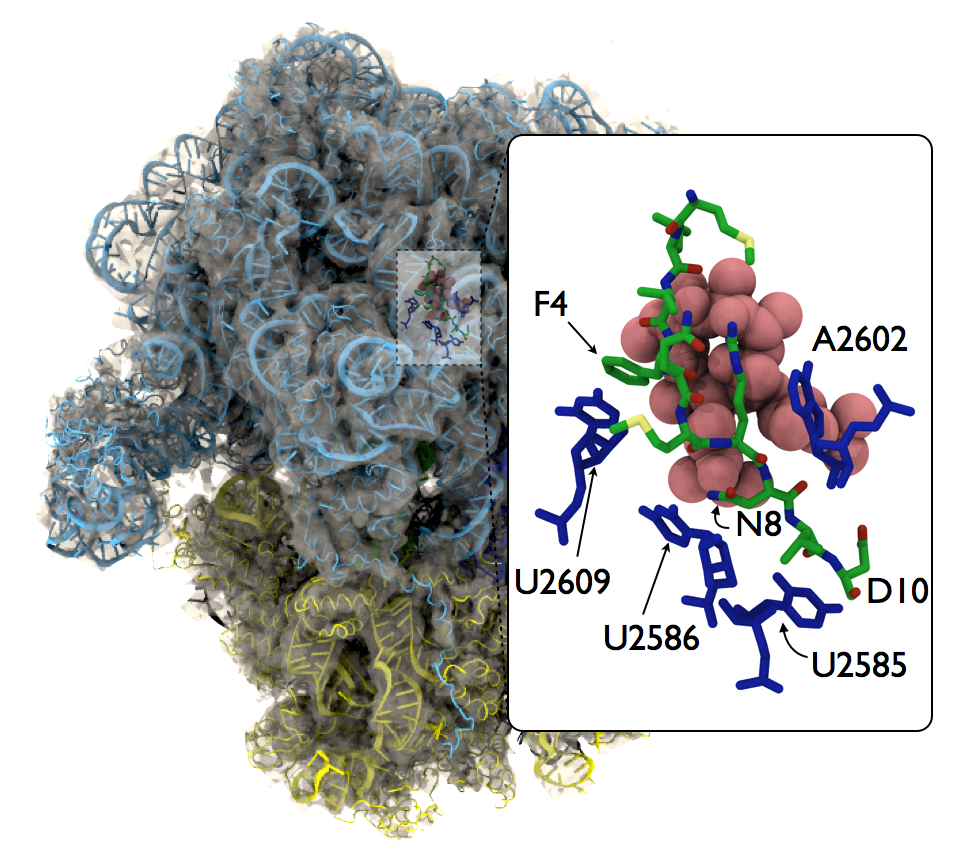
The majority of antibiotic drugs target the bacterial ribosomes. The macrolide antibiotic drug family perhaps is the most prescribed medicine used in human's history. Overuse of antibiotic drugs cause the so-called drug-resistence problem which means the drug is no more effective fighting bacterial growth. For macrolide drugs, the resistance begins with the translational stalling caused by the resistant gene-coded leard peptide. Our recent research found that the behavior of this stalling depends on the molecular structures of the drug molecules.
The macrolide telithromycin (TEL) is a representative of the newest generation of macrolide antibiotic drugs. Compared to an older dur erythromycin (ERY), TEL lacks a C3 cladinose sugar 2 which was suggested to be important structural features that make bacteria less resistant to TEL. The subtle different behaviors between ERY and TEL were exemplified by a series of biochemical experiments in our collaborator the Mankin lab, which showed that antibiotic action of ERY and TEL can be selectively turned on and off by a single mutation to the C-terminus (the 10th residue) of nascent polypeptide ErmBL: the D10-to-E10 mutant stalls with ERY, but not with TEL; the D10-to-Y10 mutant stalls with TEL, but not with ERY. With MDFF, we have resolved an all-atom structure of the complex with explicit solvent. Our molecular dynamics simulations have shown that a hydrogen-bond interaction between the C3 cladinose suger 2 and the C-terminus of ErmBL determines the stalling behavior of the translating bacterial ribosome. Read more on our paper.
Publications
Investigators
- Elizabeth Villa
- Leonardo Trabuco
- Joachim Frank
- Eduard Schreiner
- JC Gumbart
- Kwok Yan Chan
- Chris Harrison
- Bo Liu
- Rezvan Shahoei
Page created and maintained by Elizabeth Villa, Leonardo Trabuco, Eduard Schreiner, Kwok-Yan Chan and Bo Liu.
The contents of this website were reproduced in part with permission from



{kind=link}