Residue-Based Coarse Graining
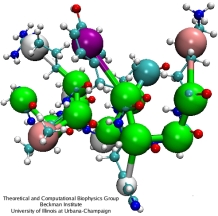
An example of coarse-grained polypeptide.
Based on the
Marrink's et. al
coarse-grained (CG) model for lipids, we have proposed a protein-lipid CG model.
Clusters of ~10 atoms (including hydrogens) are substituted by a single CG bead:
four water molecules become one ``water'' bead; an ion with its hydration shell
becomes an ``ion'' bead; chemical building blocks of lipids are reduced to single CG beads,
and each amino-acid is represented by two CG beads - one for the backbone and one for the
side-chain (except for glycine, which is represented by a single backbone CG bead).
After building the CG structure, one needs to define
rules determining its dynamics. Following a common approach in
molecular modeling, we assume that CG beads are point-like masses
that obey Newtonian mechanics, interacting through effective
potentials. Bonded beads are connected by harmonic springs, and harmonic
angular potentials help to maintain shape of molecular chains.
Long-range interaction is represented by the Lennard-Jones 6-12
potential, and also by the Coulomb potential. The bond lengths and angles
for the CG model are usually extracted from the averaging of corresponding
distances and angles over representative all-atom structures.
The developed protein-lipid CG model proved to gain a substantial speed-up in comparison
with all-atom simulations. While in the latter case the time step
is limited to 1-2 fs, our CG simulations can run with 25-50 fs time
step (because of the greater masses of the particles and smoother potentials
for their interactions). Possible speed-up depends on the computer power at hand.
For example, simulating a 300,000-particle all-atom system on 48
processors, we obtained 0.1 ns of dynamics in one day. The same system
in CG representation comprised 30,000 particles and reached performance
was 150 ns in a day, because of the smaller number of particles per
processor and larger integration time step. Accordingly, the speed-up in this case is 1500 times.
Applications of the resisude-based CG model are described in the following articles:
Publications Database
Transport-related structures and processes of the nuclear pore complex studied through molecular dynamics.
Lingling Miao and Klaus Schulten. Structure, 17:449-459, 2009.
Molecular models need to be tested: the case of a solar flares discoidal HDL model.
Amy Y. Shih, Stephen G. Sligar, and Klaus Schulten. Biophysical Journal, 94:L87-L89, 2008.
Four-scale description of membrane sculpting by BAR domains.
Anton Arkhipov, Ying Yin, and Klaus Schulten. Biophysical Journal, 95:2806-2821, 2008.
Application of residue-based and shape-based coarse graining to biomolecular simulations.
Peter L. Freddolino, Anton Arkhipov, Amy Y. Shih, Ying Yin, Zhongzhou Chen, and Klaus Schulten. In Gregory A. Voth, editor, Coarse-Graining of Condensed Phase and Biomolecular Systems, chapter 20, pp. 299-315. Chapman and Hall/CRC Press, Taylor and Francis Group, 2008.
Molecular modeling of the structural properties and formation of high-density lipoprotein particles.
Amy Y. Shih, Peter L. Freddolino, Anton Arkhipov, Stephen G. Sligar, and Klaus Schulten. In Scott Feller, editor, Current Topics in Membranes: Computational Modeling of Membrane Bilayers, chapter 11, pp. 313-342. Elsevier, 2008.
Molecular models need to be tested: the case of a solar flares discoidal HDL model.
Amy Y. Shih, Stephen G. Sligar, and Klaus Schulten. Biophysical Journal, 94:L87-L89, 2008.
Assembly of lipoprotein particles revealed by coarse-grained molecular dynamics simulations.
Amy Y. Shih, Peter L. Freddolino, Anton Arkhipov, and Klaus Schulten. Journal of Structural Biology, 157:579-592, 2007.
Assembly of lipids and proteins into lipoprotein particles.
Amy Y. Shih, Anton Arkhipov, Peter L. Freddolino, Stephen G. Sligar, and Klaus Schulten. Journal of Physical Chemistry B, 111:11095-11104, 2007.
Disassembly of nanodiscs with cholate.
Amy Y. Shih, Peter L. Freddolino, Stephen G. Sligar, and Klaus Schulten. Nano Letters, 7:1692-1696, 2007.
The role of molecular modeling in bionanotechnology.
Deyu Lu, Aleksei Aksimentiev, Amy Y. Shih, Eduardo Cruz-Chu, Peter L. Freddolino, Anton Arkhipov, and Klaus Schulten. Physical Biology, 3:S40-S53, 2006.
Coarse grained protein-lipid model with application to lipoprotein particles.
Amy Y. Shih, Anton Arkhipov, Peter L. Freddolino, and Klaus Schulten. Journal of Physical Chemistry B, 110:3674-3684, 2006.
Back to the main coarse-graining web page.
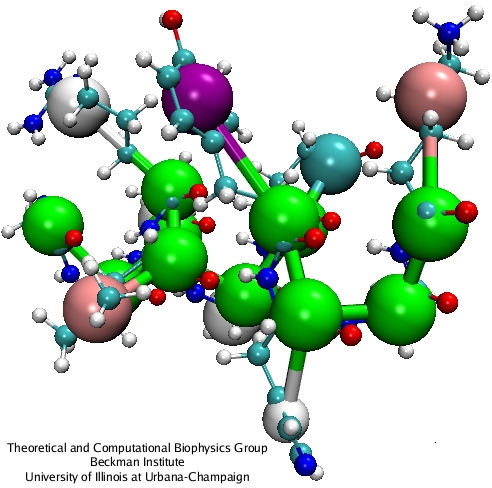 An example of coarse-grained polypeptide.
An example of coarse-grained polypeptide.


