Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001

Ever since the 1953 discovery of DNA's double helix structure researchers wondered how the double strands are separated so that genetic information stored inside the helix can be delivered from generation to generation. A class of proteins known to achieve this separation are DNA helicases (see Sept 2006 highlight), molecular motors that operate at a fork where a double stranded DNA separates into two single stranded DNAs. Helicases translocate along one of the single stranded DNAs, pushing forcefully into the fork to split the double stranded DNA apart further. Helicases seem to work, though, both through force and through persuasion, exposing to the double stranded DNA a surface that is apparently conducive for strand separation. This property suggests itself on account of the fact that many of the amino acid side groups at the relevant surface are highly conserved among species or evolved from species to species through pairwise mutation. But what chemical strategy evolution had in mind in molding the surface was not realized. Recently, however, researchers seeking artificial means of splitting double stranded DNA apart might have found a key clue. They pulled double stranded DNA at one of its single strands by means of an atomic force microscope from DNA's native salt water environment into a so-called non-polar solvent. The force - distance curve measured suggested that the DNA actually split apart, but there existed no direct experimental means of viewing the splitting. The researchers employed instead molecular dynamics simulations, using NAMD, that indeed clearly revealed the splitting of the DNA strands at a water - oil (octane) interface as reported in a recent publication. The study suggests how helicases achieve the splitting of DNA strands, namely by altering the local environment of DNA from water-like (hydrophilic) to oil-like (hydrophobic). More information here.
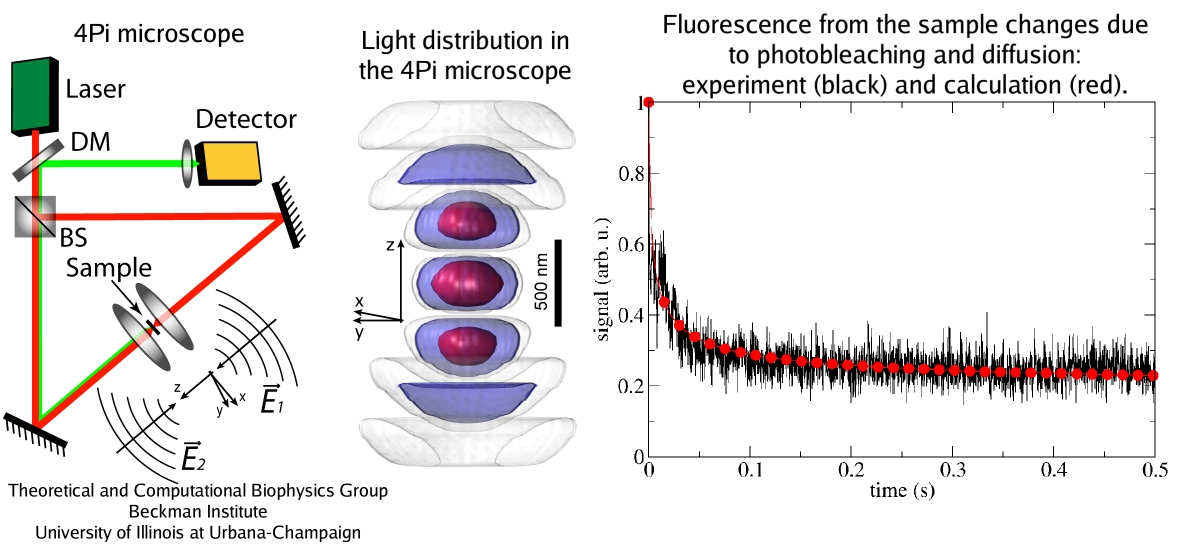
image size:
223.7KB
made with VMD
Since Leeuwenhoek introduced it to biology 300 years ago, the light microscope has brought about multiple discoveries, many achieved through improving over time the instrument's resolution. However, in 1873 Abbe recognized that the resolution has a limit, given by the wave length of light. This limit was considered absolute, until in 1992 Hell suggested a microscope that breaks the limit postulated by Abbe. This is achieved by sending coherent light through two opposing objectives, the resulting interference pattern squeezing the radiation into spots significantly smaller than the light's wavelength. This improvement has already permitted biologists to see a new level of detail in living cells. However, the pattern of light in the Hell microscope is rather complex and certain quantitative measurements require a computational analysis to take advantage of the full benefits of the instrument. Such analysis has been accomplished and validated in a recent study, the validation involving measurements on known systems. The developed numerical algorithms harness the computational power of modern processors, in particular they resort to expoiting the computational power of graphics processors (see also the Oct 2007 highlight). The new methodology combined with the new microscopes opens the avenue to unprecedented measurements in living cells. More information can be found here.
image size:
323.1KB
made with VMD
Globins are oxygen-storing proteins, vital to life. In our blood,
hemoglobins carry oxygen from our lungs to every cell in our body. In our
muscles, myoglobins keep reserves of oxygen to make sure it is available
when needed. In some plants, leghemoglobins capture oxygen molecules that
would otherwise be harmful to the production of ammonium necessary for the
plant's survival. All these globins possess an iron-containig "heme", that
grabs on to oxygen for a short time, and share the same protein
architecture, despite large variations in their sequences. Since the heme
group is buried inside a globin, scientists wondered how oxygen makes its
way inside the protein to reach it.
Exploring the motion and energetics of globins using the program NAMD researchers learned to gather data that
permitted them to visualize, utilizing the VMD software, all the pathways taken by oxygen
migrating inside whale myoglobin (see the Aug 2006 highlight and related
publication). However, when the researchers turned their attention to
the rest of the globin family to compute their oxygen pathways, they
found, on their computational spelunking trip,
something surprising. Given the conserved architecture of all globins,
they expected to see similar oxygen pathways throughout the globin family,
but they saw the opposite! Aside from a conserved pocket right at the
heme binding site, the distribution of oxygen pathways showed very little
similarity from one globin to the next. This result is described in a
recent report, which
shows that oxygen-pathways are not conserved by evolution, and that their
location is not determined by a protein's overall architecture, but rather
by its local amino acids. The researchers also learned which amino acids
are found more often than others lining oxygen pathways, recognizing that
bulky side groups are not hindering, but favoring oxygen passage. More
information can be found here.
image size:
221.8KB
made with VMD
Modern computers include a massively parallel graphics
processing unit (GPU) designed to perform geometry and lighting
calculations at blistering speeds.
State of the art GPUs can perform 0.5 teraFLOPS with their
hundred cores.
The tremendous computational power of GPUs
was untapped by scientific computations because it could only be
accessed with difficulty until now.
As
reported in the Journal of Computational Chemistry, recent
advances
allowing GPUs to be used for general purpose computing have
boosted the performance of a number of molecular modeling applications,
including
NAMD
simulations and
VMD
electrostatic potential calculations.
The accelerated versions of these
applications run five to one hundred times faster than on the best
CPU-based hardware, allowing a single desktop computer equipped
with a GPU to provide processing power equivalent
to an entire, large computing cluster.
More information on GPU acceleration of molecular modeling
applications is provided
here.
image size:
259.0KB
made with VMD
Life on Earth learned through billions of years of evolution to make do with renewable energy. Evolution discovered early on that the sun light is an excellent renewable energy source. To harvest sun light, many different types of biological solar batteries are used. One of the simplest ones is found in so-called purple bacteria. Their cells contain about hundred bulbous invaginations of the cell membrane, pointing into the cell. The spherical bulbs contain six different kinds of proteins adding up to a total of about 250 proteins. The primary function of the bulbs is to absorb sun light and electrically charge their spherical membrane, the charge being then used to synthesize fuel for the bacterium's energy needs. The structures of the individual proteins were determined earlier through X-ray crystallography. In a recent paper, a team of experimental and computational biologists report the atomic level architecture of the entire bulb. The team used atomic force microscopy on flattened bulbs to measure the height profile of the proteins, reconstructing from the data the bulbous protein assembly. The result is a detailed picture of the bulb, showing over 4000 chlorophyll molecules being held in the bulb, absorbing sun light, and transferring its energy to reaction centers that convert the sun light into an electrical membrane voltage. Indeed, the team could calculate, using quantum and thermal physics, from the architecture the amazing efficiency of the bacterial solar battery. More on chromatophores of purple bacteria can be found here.
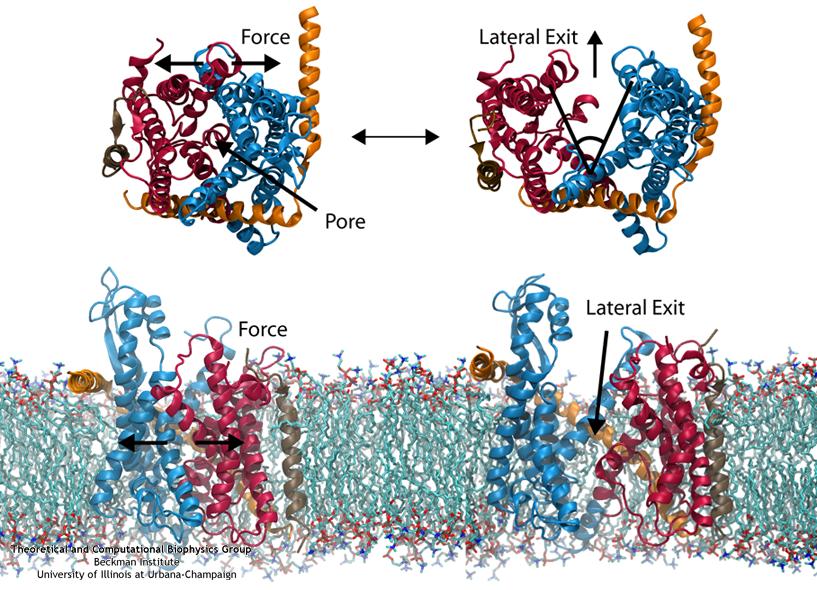
image size:
111.2KB
made with VMD
Everyone knows oil and water don't mix. Proteins observe this rule, too, some choosing to stay in the watery cytoplasm and others choosing the oily membrane. But getting into the membrane is
not easy, and most newly formed proteins require another protein, the membrane-bound translocon, to help them insert into the membrane. The translocon, surprisingly also serves as a conduit
for proteins across the membrane, thus carrying out a unique dual function. The structure of the translocon showed evidence of a likely "lateral gate", i.e., an exit from the channel into the
membrane. How the channel opened to the membrane though, and how it closed afterwards, were not clear from the structure alone. Now, molecular dynamics simulations performed with NAMD, covered in a recent publication, have permitted researchers to understand how the channel opens
laterally, how it closes, and how the oily lipids are prevented from invading the water-filled pore. Furthermore, the novel simulation technique, residue-based coarse graining, allowed the researchers to simulate the lipid-channel interactions for up to one microsecond, clearly illustrating that the
lipids do not want to mix with the channel interior. More information on these results can be found on the Protein Translocation website.
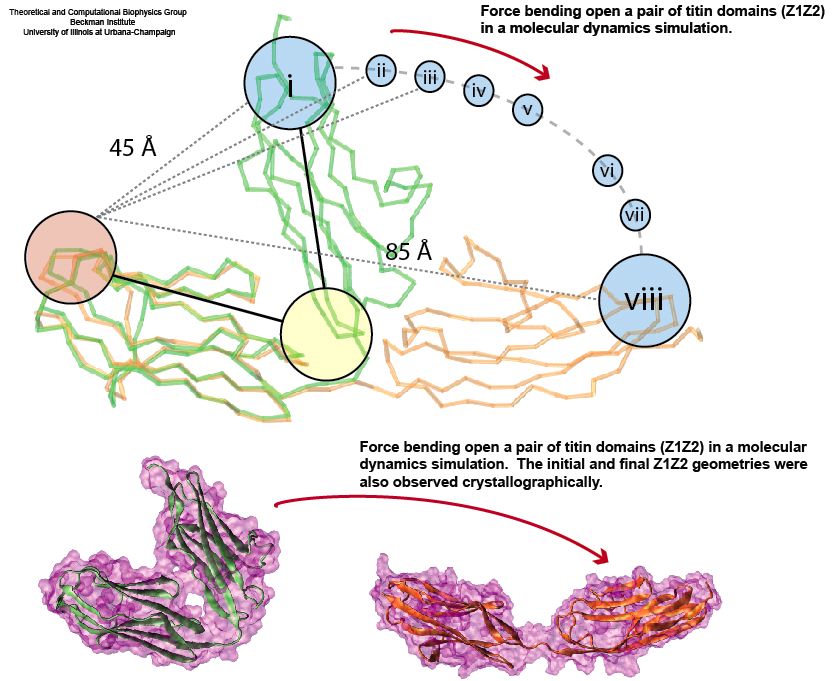
image size:
92.2KB
made with VMD
Muscle fibers, in contracting and extending, generate tremendous force that needs to be buffered to protect muscle from damage. This role falls to the protein titin, with about 27,000 amino acids the longest protein in human cells. Titin functions as a molecular rubber band, but unlike uniform rubber bands, titin is made from over 300 different protein domains strung into a chain. While experiments have found that the individual domains of titin feature remarkable resilience against mechanical stretching, little is known about the elasticity of the overall titin chain. Crystallographers teamed up with computational biologists to investigate this elasticity, focusing on two adjacent titin domains. Molecular dynamics simulations using NAMD suggest, as reported recently, that the overall elasticity of the titin chain stems in part from a zigzag, i.e., accordion-like, motion: as titin is contracted and extended, energy is stored and released in the angular tilt of adjacent domains. More on this investigation can be found here.
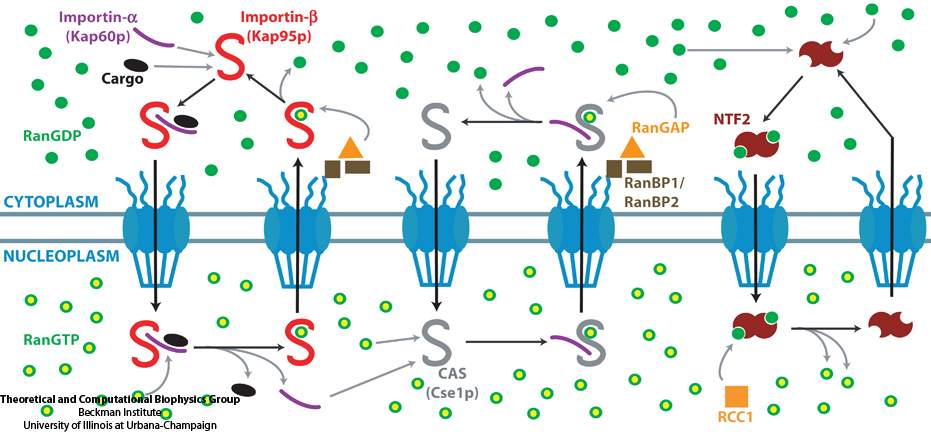
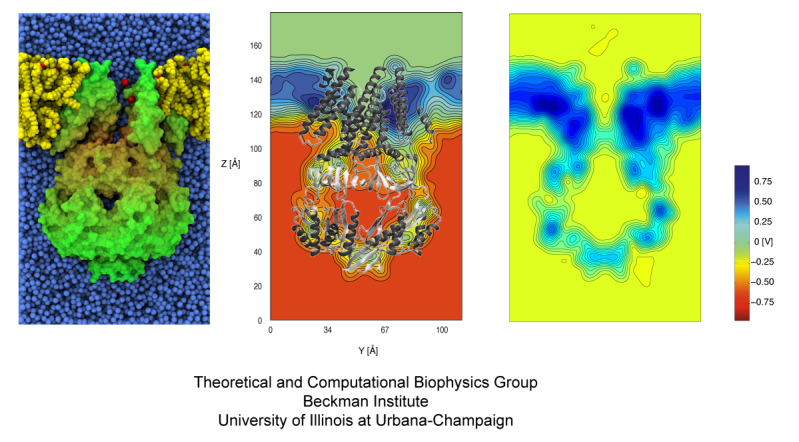
The cells of higher organisms store their genetic material, the genome, in the so-called nucleus where they organize transcription of DNA into messenger-RNA, the blueprint for proteins. The messenger-RNA leaves the cell to be decoded by ribosomes that synthesize the respective proteins. Transcription factors, also proteins, control in the nucleus which parts of the cells' genomes are transcribed. Naturally, the access to the nucleus as well as exit from it must be restricted to transcription factors and related biomolecules. This is achieved by the nuclear pores, wide channels lined with brushes of polymers. The polymers are disordered proteins and prevent passage for most cellular proteins, except for so-called transport factors which bus transcription factors, messenger RNA, and certain larger biomolecule into and out of the nucleus. How transport factors are permitted to pass the nuclear pores, despite many studies, has been largely unknown. Molecular dynamics simulations, based on relevant crystallographic structures, using NAMD provided a comprehensive picture on the passage mechanism as reported recently. The simulations, analyzed with VMD, revealed that transport factors are dotted rather regularly on their surface with spots that bind to the brushes of nuclear pore proteins. While any protein may accidentally exhibit such a binding spot or two, only transport factors offer a regular pattern of such spots on their surface that apparently is their passport permitting them movement into and out of the nucleus, i.e., helping them to glide through the pores' protein brushes. More on simulations of transport factors can be found here.
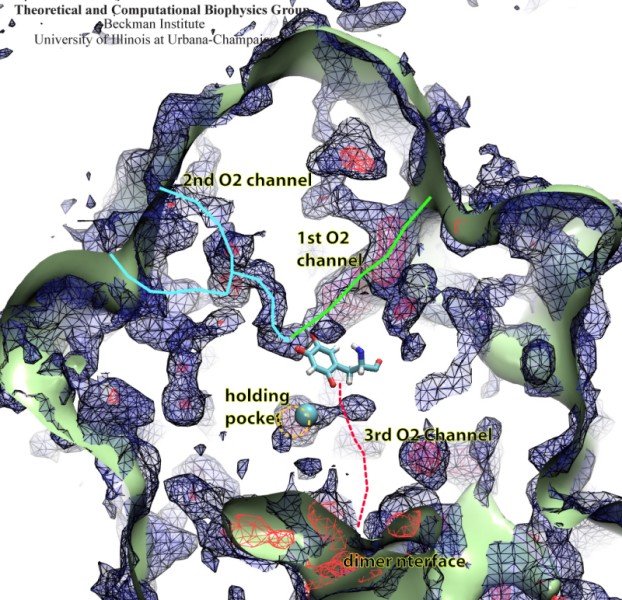
image size:
133.1KB
made with VMD
Because oxygen gas is very reactive, it is frequently employed by the cell
as a reagent by proteins called enzymes, which build the organic compounds
that the cell needs. One such enzyme belongs to the copper amine oxidase
family. These proteins transform amine-containing compounds into molecules
needed by the cell, by reacting the compounds with oxygen. Researchers
have long been interested in finding out how the various reagents reach
the buried copper active site before the final oxidation reaction can
occur. While copper amine oxidases exhibit an obvious channel for
capturing the amine compounds to be modified, it had been unclear until
now how oxygen molecules make their way through the enzyme. With the help
of computer simulations using NAMD,
researchers have identified in a recent publication, the
routes taken by oxygen inside various copper amine oxidases from different
species. In order to accomplish this, they analyzed simulations of the
motions of four copper amine oxidases, using the VMD analysis and visualization software, which
can predict the probability of finding oxygen molecules anywhere inside
the simulated proteins. This analysis found numerous oxygen conduction
routes inside each copper amine oxidase, i.e., oxygen can enter the
protein through many routes, as it would in a sponge. More information on
finding O2 migration pathways in proteins can be found here.
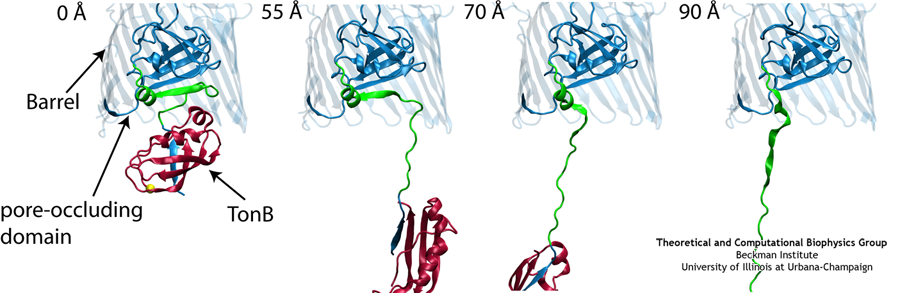
Like all organisms, bacteria have to eat. However, bringing nutrients in from the outside world is not an easy task for many bacteria that are surrounded by an extra membrane. The second
membrane, called the outer membrane, offers additional protection but at a cost: no energy can be generated or stored at the outer fringes of the cell. So, to import large, rare nutrients
that cannot cross by diffusion alone, bacteria have evolved a unique transport system which couples the inner, energy-generating membrane to the passive outer membrane, known as the
TonB-dependent transport system. TonB, an inner membrane-associated protein, transfers energy across the periplasm to a variety of outer-membrane transporters. These transporters have a
large, beta-barrel structure which is blocked in the middle by a plug called the 'luminal domain'. How TonB transfers energy to the transporter and causes the luminal domain to come out is
still a mystery though. Now with the help of computer simulations using NAMD and a recent crystal structure of TonB coupled to BtuB, the transporter responsible
for vitamin B12 transport, researchers have shown that TonB can mechanically activate the transporter by pulling on the luminal domain, causing it to leave the barrel. Using steered
molecular dynamics, it was found that TonB stayed firmly attached to the luminal domain of BtuB, even though the contact between the two is limited to just a handful of residues.
Furthermore, this pulling initiated unfolding of the luminal domain, opening a transport pathway for the substrate. These results, the subject of a recent publication and also highlighted in Science, demonstrate how a mechanical coupling can bridge the gap between the two membranes, thus
enabling outer membrane transport.
We all know sushi rolls, but just to be sure here is an easy definition: a wrapper encircles rice which holds a precious bit of fish. To make a sushi role is an
art and the same holds true for molecular sushi that is made of two lipoproteins as wrapper, lipids as rice, and membrane proteins as filling. Sushi rolls are for
eating. Molecular sushi roles are for holding membrane proteins in place for physical analysis; they actually come only in sliced form, one disc at a time. Due to
their size, the discs are called nanodiscs. Since membrane proteins are notoriously difficult to study experimentally due to their need to be in a "native"
membrane environment, nanodiscs are a great tool, furnishing a membrane environment that has been used to embed a variety of membrane proteins for biochemical
assay, including cytochrome P450's, rhodopsin, bacterial chemoreceptors, blood clotting factors, and translocation proteins. Unfortunately, it is difficult to make
either real or molecular sushi rolls (nanodiscs). In either case one needs to lay down the ingredients first. In the case of nanodiscs, one starts from the raw
ingredients which are solubilized by the detergent cholate. Removing the detergent allows the nanodiscs to self-assemble. However, the assembly process is
difficult to quantify experimentally, thus researchers rather studied the disassembly process, i.e., how detergent disassembles preformed nanodiscs. One can watch a sushi
chef make rolls, but watching the disassembly and assembly of nanodiscs is harder due the the small size. Fortunately, a computer can image the process. In a recent publication, nanodisc disassembly through the addition of increasing
concentrations of cholate was monitored through computer simulations using NAMD and verified through
experimental small-angle X-ray scattering. The study showed how cholate molecules insert themselves at the interface between the lipids and lipoproteins towards
complete disassembly. The simulations employed a new method called residue-based coarse-graining. For
more information, see our webpage on nanodiscs.
image size:
91.7KB
movie (
789.9KB
)
Mechanical forces are everywhere in human life. Strong forces power machines and cars, our body's forces let us labor and move, soft forces
are sensed through touch, even softer ones through hearing. Forces are also ubiquitous in the living cell, driving its molecular machines
and motors as well as signaling ongoing action in its surroundings. Man made, force bearing machines rely on extremely strong materials not
found in the cell. How can the cell bear substantial forces? Also, how do cells sense extremely weak forces as in hearing, surpassing most
microphones? Single molecule measurements, reviewed in a recent issue of Science, begin to answer these questions offering information on
biomolecules' mechanical responses and action. However, the information offered by these measurements is not enough to relate the
biomolecular function to the biomolecular architecture. Biomolecules in cells can move in amazing ways, but we did not know why. As
one contribution in the Science issue demonstrates,
computational modeling comes to the rescue. It can simulate the measurements and, in doing so, can reveal the physical mechanisms underlying
cellular mechanics at the atomic level. In as far as observed data are available, the simulations show impressive agreement with actual
measurements. While initially only following experiments or, at best, guiding experiments,
modeling has advanced now further and through simulated measurements
discovered on its own entirely novel mechanical properties that were later verified by experimental measurements. Experimentalists reacted to the
new competition and began to do simulations themselves. More here.
image size: 81.4KB
The five human senses are based on amazingly sensitive molecular processes: smell and taste are based on molecular recognition, hearing and touch on molecular mechanics, vision on molecular electronic excitation. Some animals have additional sensory capabilities; for example, some possess a magnetic sense used for orientation by means of the geomagnetic field. The magnetic sense has long been poorly understood since the underlying molecular process could not be identified, but recently some progress has been made. Surprisingly, animal vision has been implicated and evidence has been accumulated that animals can see the geomagnetic field. A long-hidden receptor in the eye, a protein aptly called cryptochrome, is likely involved. Unfortunately, cryptochrome exists only in minute amounts in animal eyes, e.g., those of migratory birds, so that only behavioral measurements on animals can be taken, but not physical measurements directly on cryptochromes. Fortunately, cryptochromes exist also in plants where they control hypocotyl growth inhibition in seedlings. Experimentalists have observed that cryptochrome-dependent responses in Arabidopsis thaliana seedlings are magnetic-field-dependent. Researchers have now also computationally demonstrated that light activation of plant cryptochrome is magnetic-field-dependent. A recent report showed that light excitation leads to cascading electron transfer in which electron spins are influenced by weak magnetic fields; the spin dynamics was found to influence the activation of cryptochrome. Arabidopsis thaliana cryptochrome can be produced in quantities large enough for physical measurements so that the door is now wide open for cracking the secret behind the long-mysterious magnetic sense of animals. More on our cryptochrome web site.
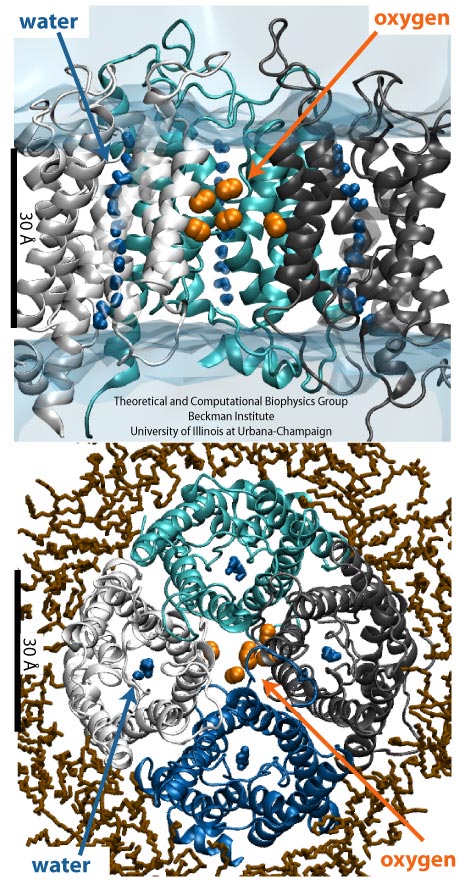
image size:
192.3KB
made with VMD
Every morning, many people drive to work, while others may bike, take the
bus or the metro.
Similarly, various biomolecules in the human body also reach their
destinations in diverse manners.
For example, to cross the cellular membrane, small hydrophobic gas molecules diffuse through the lipid
bilayer, while water molecules pass through specialized channel proteins named
aquaporins (AQPs).
Interestingly, just like one may get to work both by bus and by
driving, it has been found recently that some gas molecules may have more
than one way to cross the membrane, i.e., besides diffusion through
lipids, oxygen and carbon dioxide may also pass through AQPs.
However, the pathways that these gas molecules take remained elusive.
Using molecular dynamics performed with
NAMD, researchers have investigated the gas
permeability of AQP1 in a recent study with
two complementary methods (explicit gas diffusion simulation and implicit ligand
sampling). The simulation results suggest that while the four
monomeric pores of AQP1 function as water channels,
the central pore of AQP1 may serve as a pathway for gas molecules to
cross the membrane. More information on the simulations can be found on
the aquaporin web page.
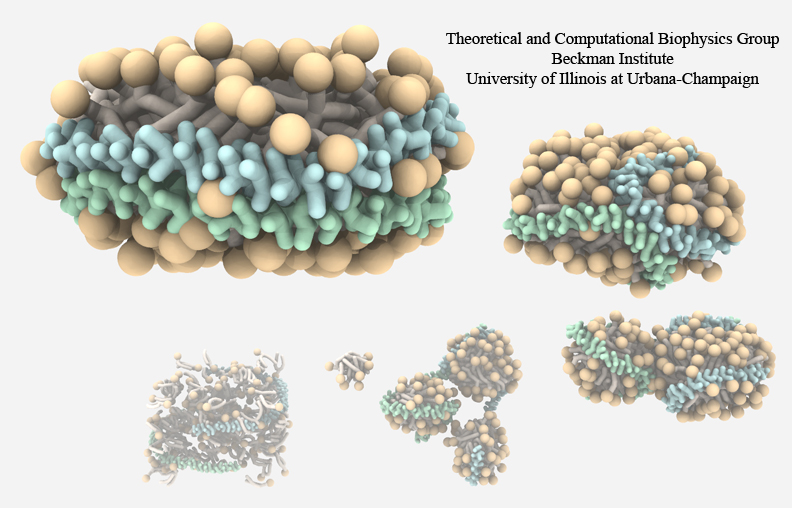
High-density lipoproteins, otherwise known as the "good cholesterol", are the body's way of naturally removing
cholesterol in the blood stream.
Since lipid and cholesterol molecules are not soluble in blood, lipoproteins are needed to collect and transport
them.
The proteins wrap themselves around the hydrophobic portions of lipids and cholesterol, effectively shielding
them from the aqueous environment and allowing them to be transported through the bloodstream to the liver for
degradation.
High-density lipoproteins exhibit a variety of shapes and sizes and presently cannot be imaged through experimental
observations.
Computational methods, however, can provide detailed images of high-density lipoprotein particles, even showing how these
particles form in the body.
As recently reported (article 1, article 2), so-called coarse-grained
molecular dynamics simulations using NAMD discovered how lipid molecules are
corralled by lipoproteins to form disc-like high-density lipoprotein particles.
The simulations show in remarkable detail the aggregation of proteins and lipids, starting from a random arrangement of lipids
that are mopped-up by two lipoproteins, eventually forcing the lipids into a disc-shape surrounded on its circumference by
belt-like lipoproteins.
For more information, see our webpages on HDL & nanodiscs and coarse-grained modeling.
Bacteria employ membrane proteins as crucial safety valves that release water and small solutes
under challenging osmotic conditions (see the
May 2006 highlight,
"Electrical Safety Valve" and the Nov 2004
highlight, "Japanese Lantern Protein"). There are valves for balancing small pressure differences between
the inside and outside of bacterial cells, that open and close readily, but there are also
ones for protection against large pressure differences as a safety measure of last resort. The
valves for balancing small pressure differences, like the one shown in the figure, include
a filter that presumably keeps the most valuable molecules inside the cell interior, though
this is not understood yet in detail. To reveal the
function of such channels a combination of X-ray crystallography, physiological measurements, and molecular
dynamics simulations using NAMD has been employed.
Crystallography, in a prior study, captured the channel in a half-way open state. Now a team of physiologists and
modelers reported the details on valve opening and closing.
The experiments, using a pipette small enough to measure currents from a single channel, MscS, along with the simulations
revealed that the channel conducts both positive and negative ions when subjected to tension and voltage. The unprecedented
comparison of experimental and computational results open a new era of quantitative cell biology that borrows
successful research strategies from physics (more on our MscS website).
image sizes:
419.0KB
&
83.2KB
made with VMD
The nucleus is responsible for storing the genome of eukaryotic cells,
isolating it from the cellular cytoplasm. Partitioning the genetic
material is very important in protecting it from cellular processes or
foreign molecules. However, the nucleus also needs to provide access
for the rest of the cell to the information stored in the genome. Numerous
nuclear pores in the nuclear envelope offer communication pathways between
the nucleoplasm and cytoplasm. The pathways are restricted to so-called
transport receptors, proteins that taxi molecules into and out of the
nucleus. If a molecule wishes to enter or leave the nucleus, it associates
with a transport receptor. The complex passes through the pore and then
dissociates. The question is why transport receptors can pass the nuclear
pores while other proteins cannot. The answer lies in the role of
FG-repeat proteins lining the pores and filling much of their free volume.
These proteins are disordered peptides, consisting of repeating
phenylalanine-glycine (FG) residues separated by a sequence of hydrophilic
linker residues. Only proteins that interact favorably with the FG-repeat
regions can pass through, while other proteins are excluded. A
recent
report used molecular dynamics via NAMD
to examine the way in which the transport factor NTF2 interacts with the
FG-repeats. The study described binding spots for FG-repeat peptides on
the surface of NTF2, confirming known binding spots discovered previously
via experimental means, and suggesting the existence of further binding
spots. The new binding spots may play a role in steering NTF2, upon import
or export, along a particular path through the nuclear pore. See also a
previous highlight from
January 2006, "Gateway to the Nucleus", as well as our
webpage on the nuclear pore complex.
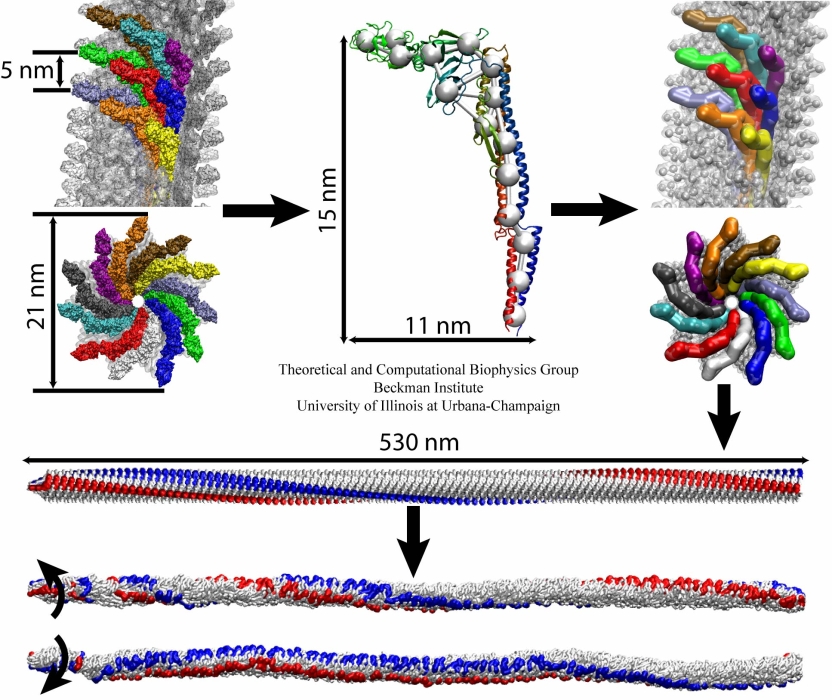
image size:
299.9KB
made with VMD
The bacterial flagellum is a large biomolecular assembly used by many types of bacteria as a helical propeller for forward swimming and turning. The flagellum is remarkable in that its properties differ greatly depending on the direction in which it is rotated, allowing the bacterium to switch between swimming straight ("running") and turning ("tumbling"). The mechanics of the flagellum are of interest both to biologists and mechanical engineers. The molecular mechanisms of the transition in the flagellum between running and tumbling modes is unknown. Because of the flagellum's size (several micrometers in length) and composition (made up of 30,000 protein subunits) it presents a challenge to computational modeling. Researchers have now achieved an advance describing the flagellum in both its running and tumbling state. For this purpose, the researchers developed a computational model of the system that glosses over atomic level detail, but resolves the shapes of all proteins making up a bacterial flagellum, simulating a simplified version of the system using the program NAMD. The results, reported recently, showed that the flagellum's transition between swimming straight and tumbling is triggered by friction due to the water around the bacterium. More information on the flagellum project can be found here.
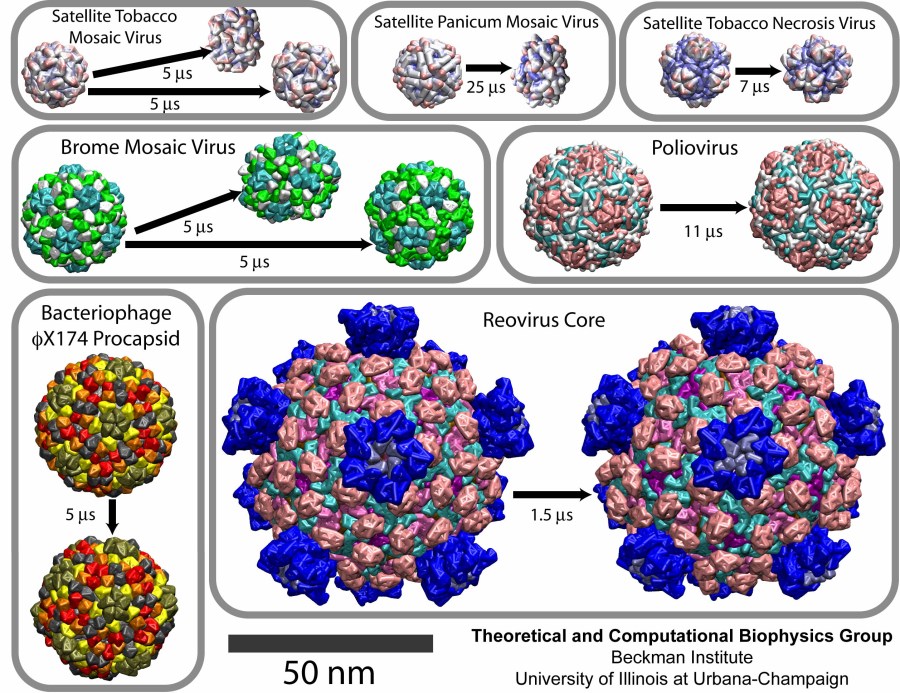
image size:
219.5KB
made with VMD
Viruses are the cause of many human diseases, from the common cold to
AIDS, and medicine is continuously searching for better ways to
battle viruses through vaccination or medication. Detailed knowledge
of the life cycles of viruses should be useful in the treatment of
viral diseases. A key focus of investigations is the virus capsid, a
protein coat that protects the viral genome, but also triggers
release of the genome and other viral factors upon contact with the
body's cells. X-ray crystallography has resolved the average
structures of many types of virus capsids, providing the basis for
detailed investigations, for example by means of molecular dynamics
methods, of capsid dynamical properties, e.g., in assembly and
disassembly. Unfortunately, due to their large size most virus
capsids are beyond the reach of molecular dynamics simulations, with
one notable exception
(see the March 2006
highlight "Simulating an Entire Life Form"). This earlier simulation
allowed researchers to develop and test a method for coarse-grained
molecular dynamics simulations that glosses over atomic detail and,
thereby, permits microsecond descriptions of entire viral particles. As reported recently
(see also journal cover)
such simulations, employing the program NAMD,
were applied to the empty capsids of several viruses. These simulations
revealed a variety of behaviors, from rapid collapse to high stability,
depending on the strength of interactions between the proteins from which
capsids are built. The new method offers unprecedented views of capsid
dynamics that may assist in battling viral diseases. More information on
the simulations can be found on our virus web page.



{kind=link}