ATP hydrolysis in F1-ATPase
Why is F1Fo-ATP synthase so important?
F1Fo-ATP synthase, or ATP synthase for short, is one of the most abundant proteins in every organism. It is responsible for synthesizing the molecule adenosine tri-phosphate (ATP), the cells’ energy currency. ATP is depicted in Fig. 1 and used to power and sustain virtually all cellular processes needed to survive and reproduce. Even when at rest, the human body metabolizes more than half its body weight in ATP per day, this figure rising to many times the body weight under conditions of physical activity.
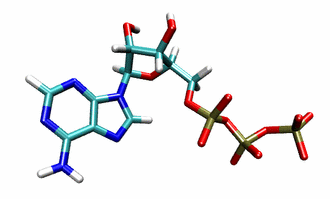 Figure 1: Molecular view of ATP.
Figure 1: Molecular view of ATP.
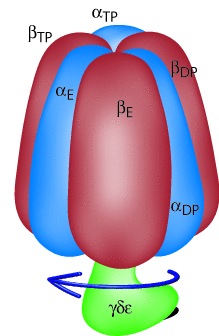 Figure 2: Schematic view of F1-ATPase. The individual subunits
are labeled with greek letters and the arrow indicates the direction
of rotation in the hydrolysis mode.
Figure 2: Schematic view of F1-ATPase. The individual subunits
are labeled with greek letters and the arrow indicates the direction
of rotation in the hydrolysis mode.
What do we know about F1Fo-ATP synthase?
Researchers have been trying to uncover the "secret" behind ATP synthase’s very efficient mode of operation for quite some time. Unfortunately, even after more than 30 years of study, we still don’t fully understand how F1Fo-ATPase really works. The protein consists of two coupled rotary molecular motors, called Fo and F1, respectively, the first one being membrane embedded and the latter one being solvent exposed.
One of the most important breakthroughs in the field was the determination of an atomic resolution X-ray crystal structure for the F1 part of ATP synthase. This allowed researchers, for the first time, to connect biochemical data to the three dimensional structure of the protein (Abrahams et al., Nature 370:621-628, 1994). The X-ray structure beautifully supported Paul Boyer’s "binding change mechanism" (Boyer, Bioch. Bioph. Acta 215-250, 1993) as the modus operandi for ATP synthase’s rotational catalytic cycle and lead to the 1997 Nobel Prize in chemistry for Boyer and Walker.
F1-ATPase in its simplest prokaryotic form (shown schematically in Fig. 2) consists of a hexameric assembly of alternating α and β subunits arranged in the shape of an orange. The central cavity of the hexamer is occupied by the central stalk formed by subunits γ, δ and ε. Due to a lack of high resolution structures for the Fo part of ATP synthase, much less is known about this subunit. It is currently thought that a transmembrane proton gradient drives rotation of the c-subunit ring of Fo which is then coupled to movement of the central stalk. The rotation of the latter eventually causes conformational changes in the catalytic sites located in F1 leading to the synthesis of ATP.
What are some of the missing pieces in our understanding of F1?
ATP synthase can be separated into its two constituent subunits F1 and Fo, which can then be studied individually. Solvated F1 is able to hydrolyze ATP and experiments pioneered by Noji et al. (Nature 386:299-302, 1997) have shown that ATP hydrolysis in F1 drives rotation of the central stalk. However, we don’t know if ATP hydrolysis itself or rather binding of ATP to the catalytic sites induces rotation. We would also like to know how the binding pockets cooperate during steady-state ATP hydrolysis to achieve their physiological catalysis rates. It has been suggested that ATP binding and product unbinding provide the main "power stroke" and that the actual catalytic step inside the binding pockets is equi-energetic, but, unfortunately, there is currently no consensus regarding this issue. In any case, since ATP in solution is a very stable molecule, the catalytic sites have to be able to lower the reaction barrier toward product formation considerably in order to cause efficient hydrolysis.
 Figure 3: Reactant state conformation of ATP hydrolysis in the
catalytic site of F1-ATPase.
Figure 3: Reactant state conformation of ATP hydrolysis in the
catalytic site of F1-ATPase.
Computational Study of ATP hydrolysis in F1-ATPase
Our research focuses on investigating the ATP hydrolysis reaction and its interaction with the protein environment in the catalytic sites of F1-ATPase using computer simulations. To be able to study a chemical reaction inside the extended protein environment provided by the catalytic sites we employ combined quantum mechanical/molecular mechanical (QM/MM) simulations to investigate both the βTP and βDP catalytic sites. Fig. 3 depicts the quantum mechanically treated region of the former. Quite surprisingly, our simulations show that there is a dramatic change in the reaction energetics in going from βTP (strongly endothermic) to βDP (approximately equi-energetic), despite the fact that the overall protein conformation is quite similar. In both βTP and βDP, the actual chemical reaction proceeds via a multi-center proton relay mechanism involving two water molecules. A careful study of the electrostatic interactions between the protein environment and the catalytic core region as well as several computational mutation studies identified the "arginine finger" residue αR373 as the most significant element involved in this change in energetics.
Several important conclusions can be drawn from our simulations: Efficient catalysis proceeds via a multi-center proton pathway and a major factor for ATPase’s efficiency is, therefore, the ability to provide the proper solvent environment by means of its catalytic binding pocket. Furthermore, the sidechain of the arginine finger residue αR373 is found to be a major element in signaling between catalytic sites to enforce cooperation since it controls the reaction barrier height as well as the reaction equilibrium of the ATP hydrolysis/synthesis reaction.
Computational Study of ATP hydrolysis in V1-ATPase
Given that ATP is the universal carrier of energy in all living systems, ATP synthase serves as a ubiquitous, evolutionary conserved enzyme, present in bacterial cell membranes, thylakoid membranes of chloroplasts, as well as the inner membrane of mitochondria. A second class of ATPases, vacoular or V-ATPases, are thought to have originated from an ancestral enzyme in common with FATPases. The main function of V-ATPases in eukaryotes is to transport protons across the membrane by using the energy produced by ATP hydrolysis. V-ATPases also catalyze ATP synthesis, exploiting the energy of proton flow in certain eubacteria, such as Thermus thermophilus.
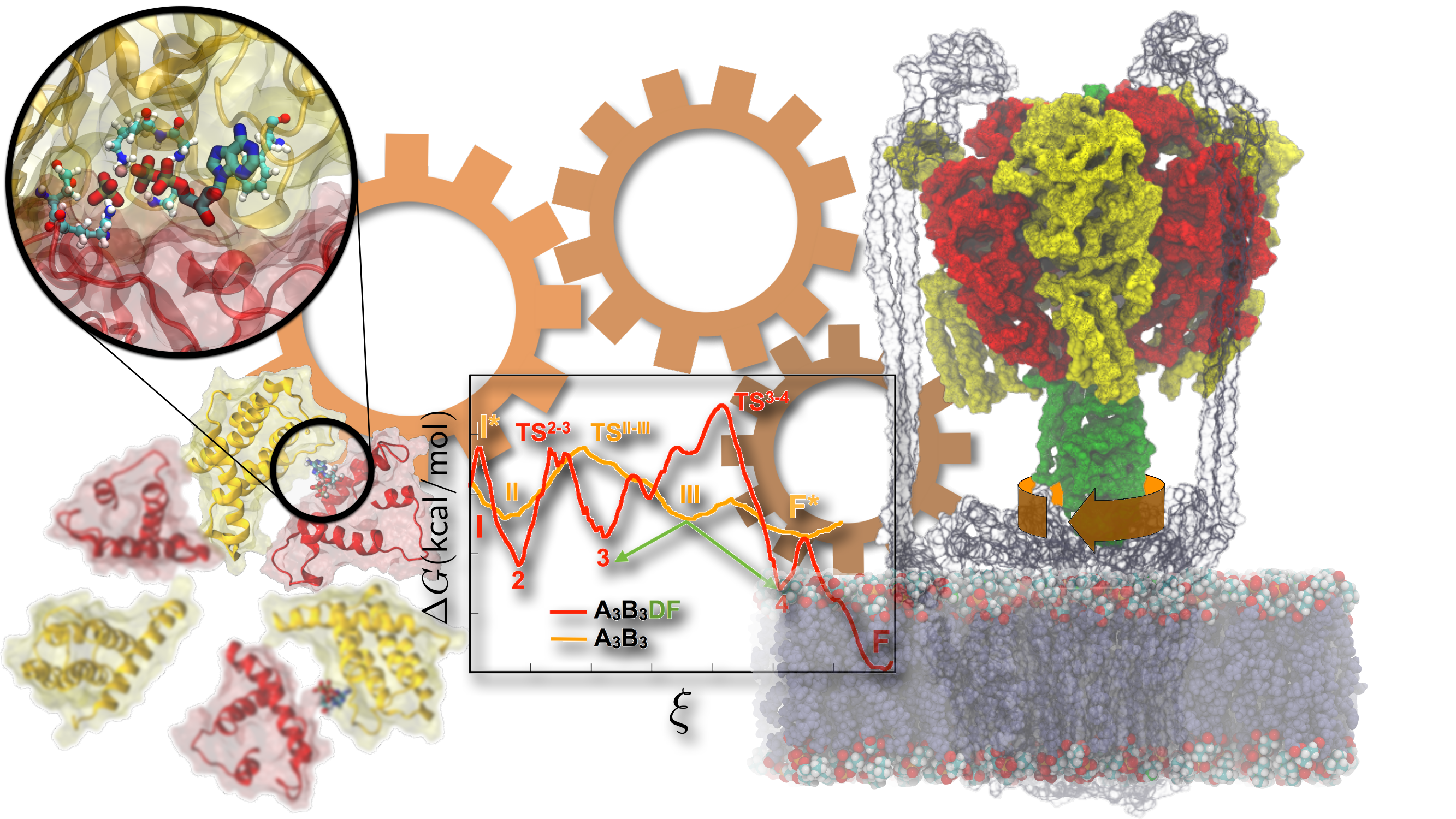
Notwithstanding the wealth of available biochemical and structural information inferred from years of experiments, the precise molecular mechanism whereby vacuolar (V-type) ATP synthase fulfills its biological function remains largely fragmentary. Recently, crystallographers provided the first high-resolution view of ATP activity in Enterococcus hirae V1-ATPase. Employing a combination of transition-path sampling and high-performance free-energy methods, the sequence of conformational transitions involved in a functional cycle accompanying ATP hydrolysis has been investigated in unprecedented detail over an aggregate simulation time of 65 microseconds (Fig. 4). Our simulated pathways reveal that the chemical energy produced by ATP hydrolysis is harnessed via the concerted motion of the protein-protein interfaces in the V1-ring, and is nearly entirely consumed in the rotation of the central stalk. Surprisingly, in an ATPase devoid of a central stalk, the interfaces of this ring are perfectly designed for inducing ATP hydrolysis. However, in a complete V1-ATPase, the mechanical property of the central stalk is a key determinant of the rate of ATP turnover. The simulations further unveil a sequence of events, whereby unbinding of the hydrolysis product (ADP + Pi) is followed by ATP uptake, which, in turn, leads to the torque generation step and rotation of the center stalk. Molecular trajectories also bring to light multiple intermediates, two of which have been isolated in independent crystallography experiments.
|
|
|
|
Figure 4: Free energy calculations demonstrate the mechanism of energy storage and motor-action in V-type ATPases. |
Publications
Investigators
Other QM/MM projects
Acknowledgment
This material is based upon work supported by the National Science Foundation under Grant No. MCB1616590, 0234938 and National Institute of Health Grant No. R01-GM067887-11. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.


