Research Projects - Other
The nature of modern science is that it is ever-changing, energetically crossing boundaries heretofore defined by traditional areas of inquiry. Research at the Theoretical and Computational Biophysics group reflects this dynamic, with studies employing theoretical perspectives and methodological approaches or addressing topics that don't fall easily into one of the above categories. Included in this broad category are studies of a four-way DNA junction, the nuclear pore complex, gas transport in hydrogenase that may provide a source of renewable fuel, and other topics.
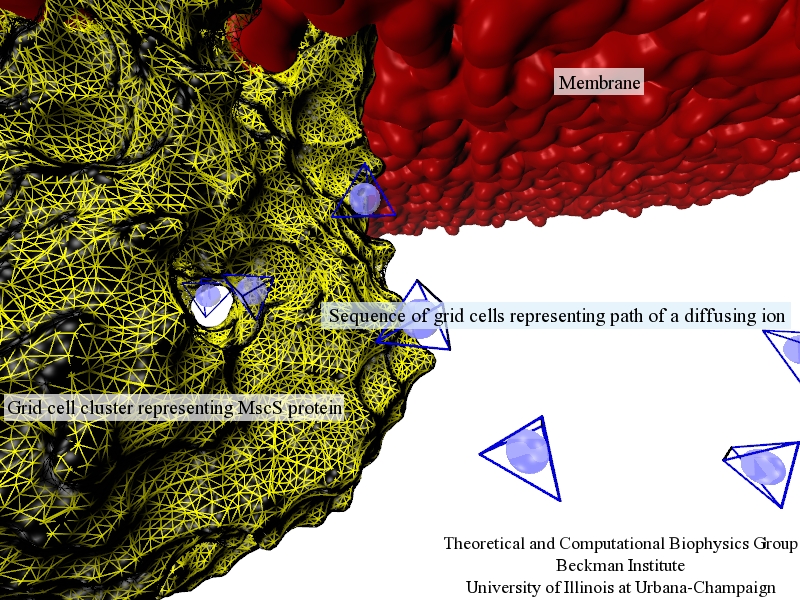
image size:
411.0KB
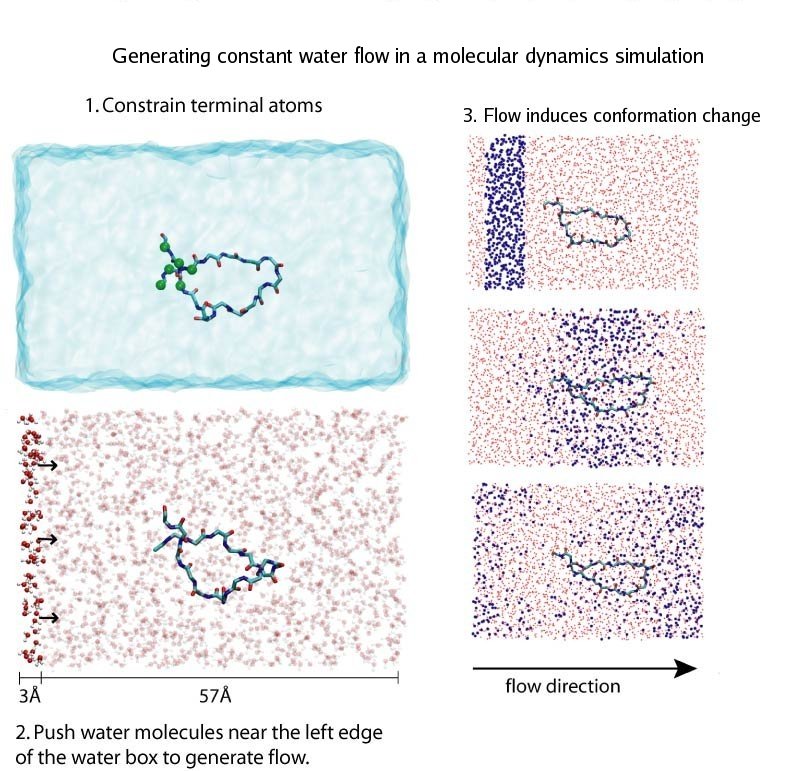
Traffic flow in a city is affected by large-scale features, like the layout of road networks, as much as it is by small-scale ones, like traffic lights at a road junction. Likewise, the transport of small molecules in cells occur on multiple scales. For example, ions diffusing through the mechanosensitive channel of small conductance (MscS) (see highlights from Jul 2011, "Smart Bacterial Safety Valve", Mar 2008, "Observation and Simulation depict Cell's Safety Valve", Feb 2007, "Observing and Modeling a crucial Membrane Channel", May 2006, "Electrical Safety Valve", and Nov 2004, "Japanese Lantern Protein") must navigate the intricate geometry of the MscS, which varies by the Ångstrom. At the same time, the distribution of ions within hundreds of Ångstroms of the MscS fluctuate as ions escape through the channel, thus changing the electrostatic landscape seen by other ions as they approach the MscS. In order to model both the fine and bulk aspects of diffusion in systems like that of the MscS, scientists have proposed, in a recent report, a method that marries the high spatial resolution of molecular dynamics to long range diffusion. In the new description, biomolecules "diffuse" by hopping through a grid under the influence of Coulomb and other forces. More on our Kinetic Diffusion web site.

image size:
223.7KB
made with VMD
image size:
323.1KB
made with VMD
image size:
221.8KB
made with VMD
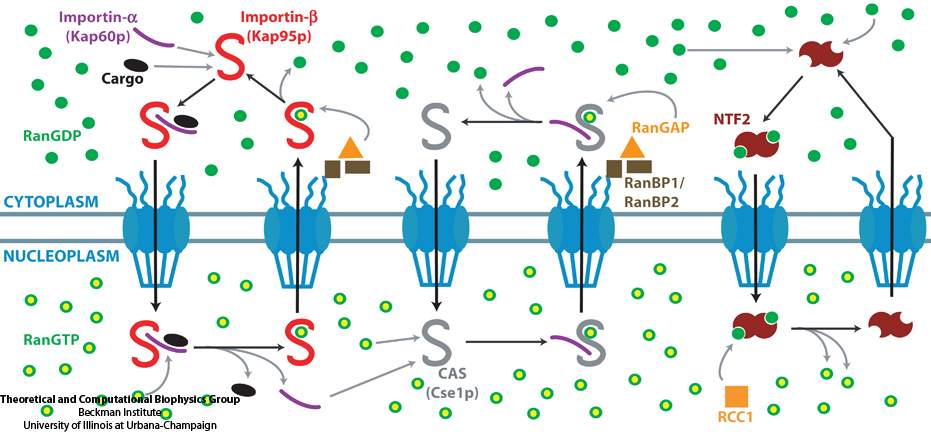
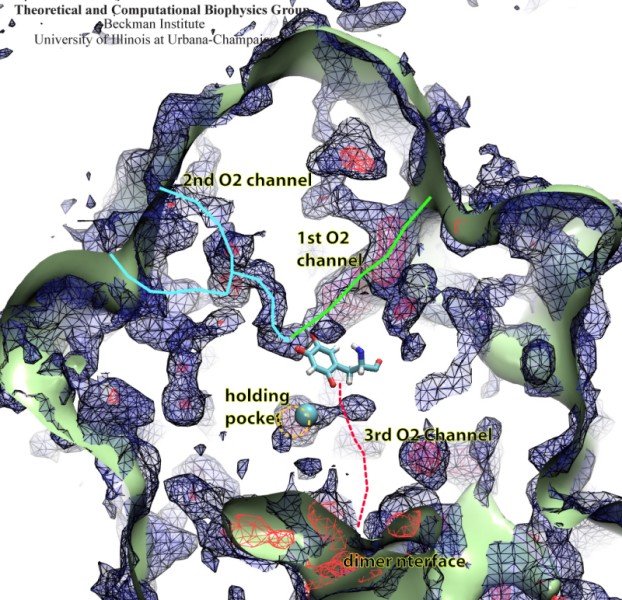
image size:
133.1KB
made with VMD
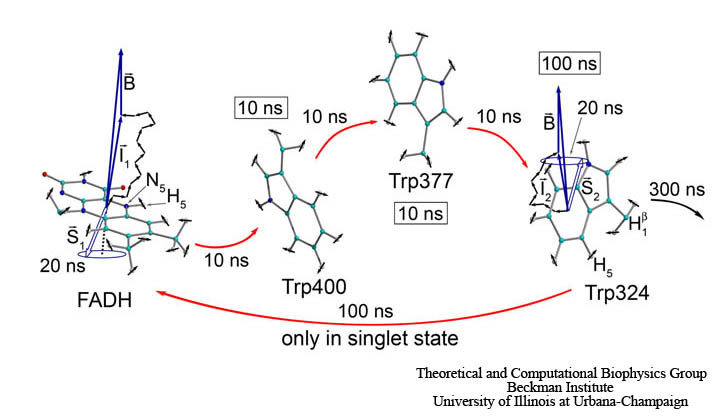
image size: 81.4KB
image sizes:
419.0KB
&
83.2KB
made with VMD
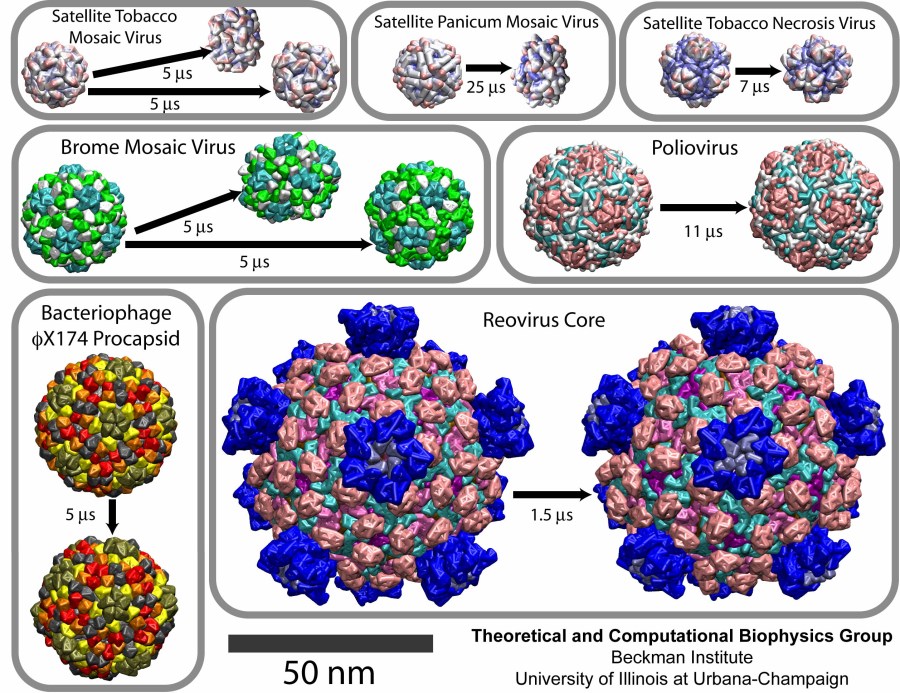
image size:
219.5KB
made with VMD
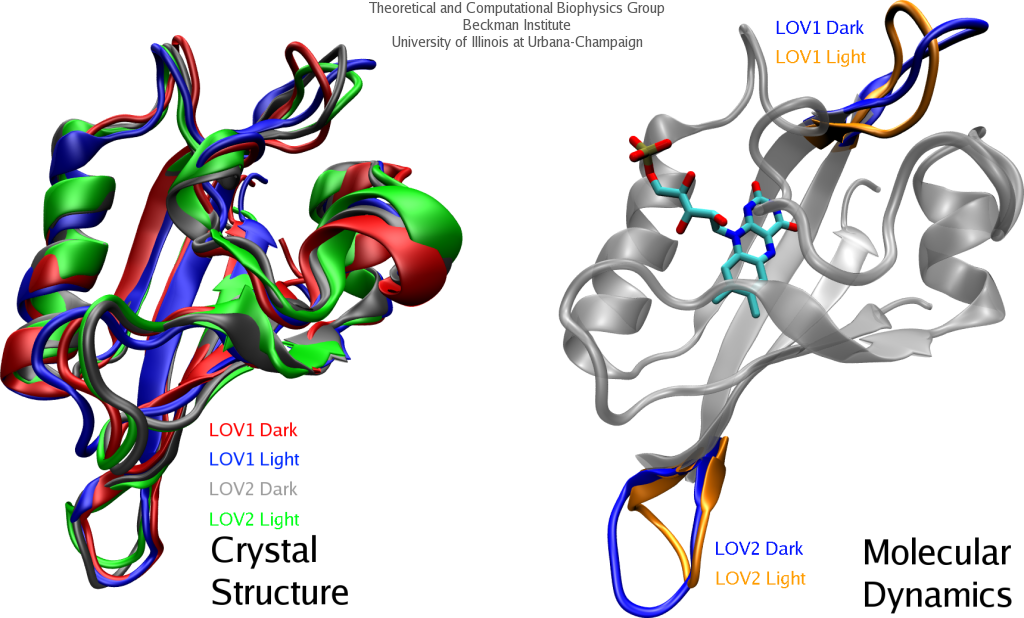
image size:
397.0KB
made using VMD
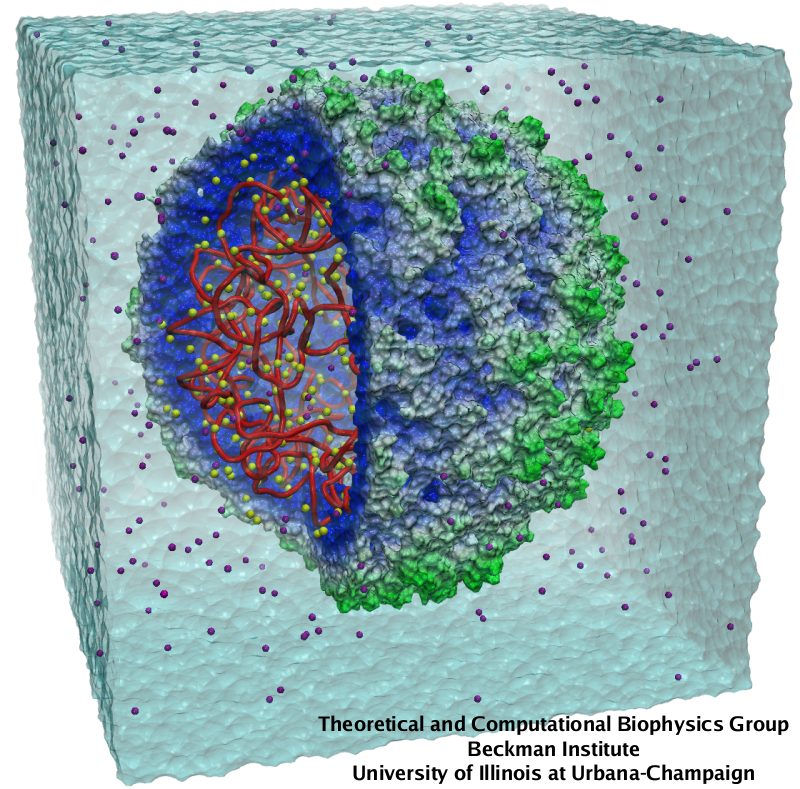
image size:
150.4KB
made with VMD
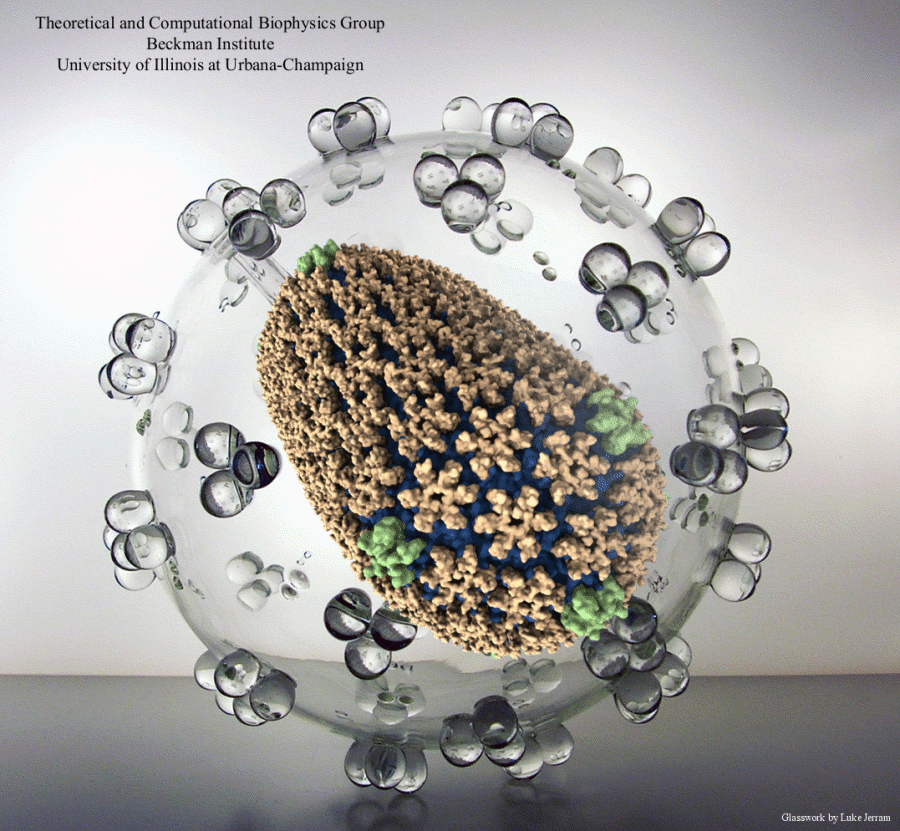
Viruses, the cause of many diseases, are the smallest natural organisms known. They are extremely primitive and parasitic such that biologists refer to them as "particles", rather than organisms. Viruses contain in a protein shell, the capsid, their own building plan, the genome, in the form of DNA or RNA. Viruses hijack a biological cell and make it produce from one virus many new ones. Viruses have evolved elaborate mechanisms to infect host cells, to to produce and assemble their own components, and to leave the host cell when it bursts from viral overcrowding. Because of their simplicity and small size, computational biologists selected a virus for their first attempt to reverse engineer in a computer program, NAMD, an entire life form, choosing one of the tiniest viruses for this purpose, the satellite tobacco mosaic virus. As described in a recent report, the researchers simulated the virus in a small drop of salt water, altogether involving over a million atoms. This provided an unprecedented view into the dynamics of the virus for a very brief time, revealing nevertheless the key physical properties of the viral particle as well as providing crucial information on its assembly. It may take still a long time to simulate a dog wagging its tail in the computer, but a big first step has been taken to simulate living organisms. Naturally, this step will assist modern medicine (more on our satellite tobacco mosaic virus web page).
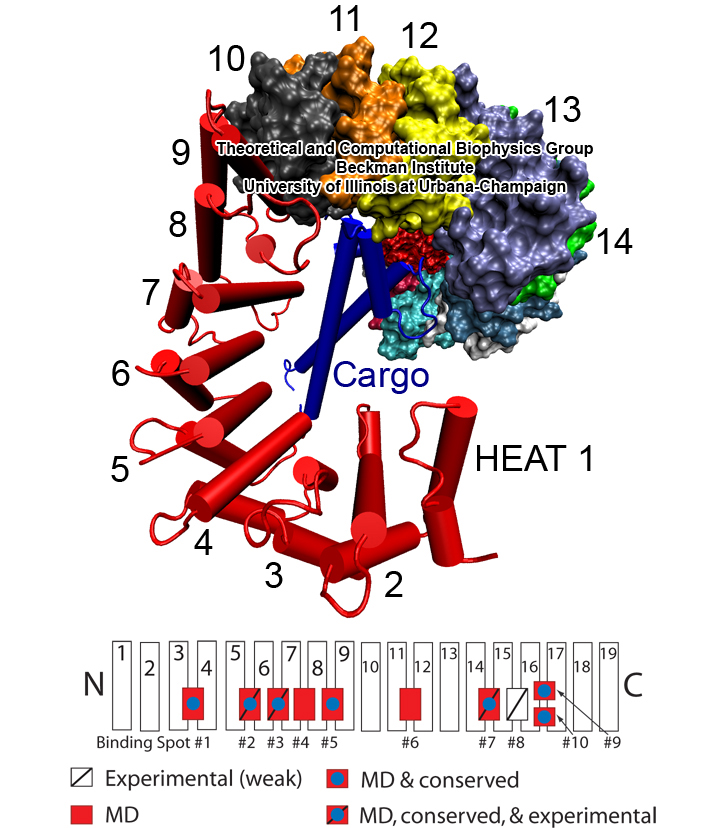
image size:
360.8KB
made with VMD
Eukaryotic cells envelop their genetic material in the cell nucleus whose boundary contains numerous pores. Only small molecules can pass through these nuclear pores unhindered. For all larger ones, passage is highly selective and controlled. The control involves import and export proteins (transport receptors) that load and release cargo on the proper side of the nucleus upon interaction with signaling proteins. Researchers are presently solving the structure of the nuclear pore and its transport receptors with increasing resolution, and the first atomic level investigation into the mechanism of nuclear pore selectivity has recently been reported [paper]. The study inspected the interaction between the transport receptor importin-β with key nuclear pore proteins that appear disordered near the center of the pore and contain characteristic phenylalanine-glycine sequence repeats. Molecular dynamics simulations using NAMD and analyzed using VMD revealed a key insight into the selectivity mechanism. The simulations showed that the key sequences of the repeat proteins interact strongly with certain spots on the surface of importin-β. The study confirmed spots that had previously been identified experimentally and, moreover, found numerous binding spots not yet seen in experiment. Further experiments and simulations promise an understanding of the selectivity of entry and exit from the nucleus, a key element of the cell's genetic control. For more information see our nuclear pore complex webpage.
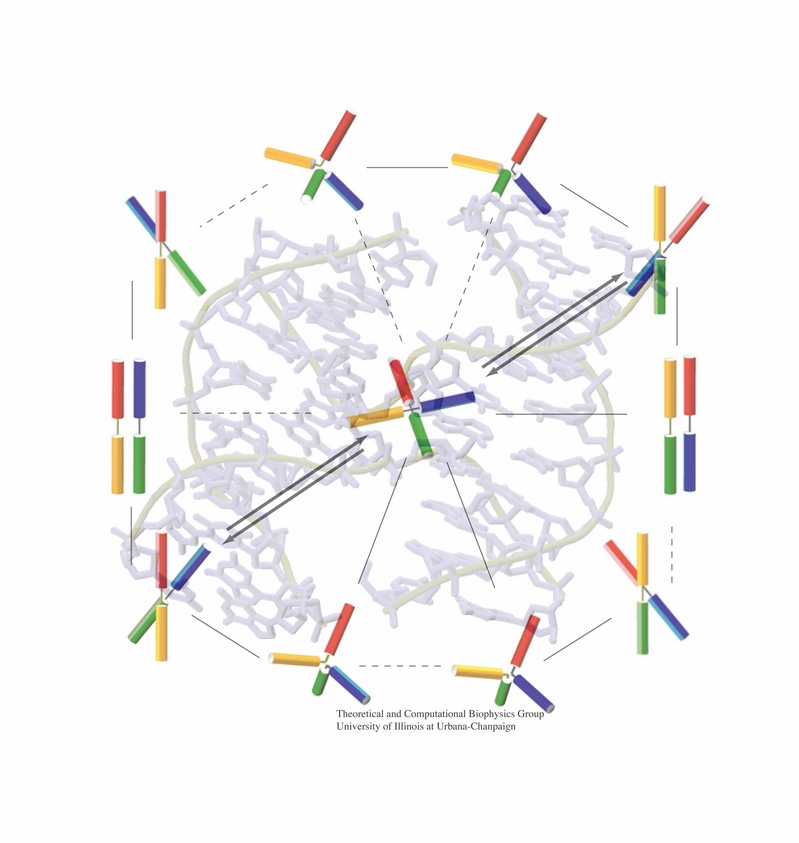
image size:
199.4KB
made with VMD
An important means for generating genetic diversity to provide raw material for evolution and maintain genomic stability is sexual reproduction. At the molecular level, the genes of two individuals are mixed through a process called homologous recombination. This process is found also in many simple life forms, even bacteria. At the beginning of recombination, two DNA duplexes, e.g., from mother and father, are aligned next to one another as the result of homology search, i.e., like strands are brought together with like strands. The four single DNA strands, two in each duplex, cross reciprocally two of the strands between the duplexes. The result is a joint molecule that contains DNA crossovers, named Holliday junctions. The Holliday junction is highly polymorphic in moving along two DNA duplexes, exchanging their DNA. Researchers are now investigating the physical mechanism of Holliday junction migration. The polymorphic, dynamic character of this migration makes observations difficult and the researchers resorted to molecular dynamics simulations using NAMD. The results, reported recently, resolved the dynamics of maternal-paternal DNA exchange through Holliday junction transitions in unprecedented detail providing an atomic level view of sexual reproduction. Check a brief review on our website.
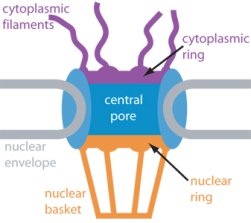
image size: 18.0KB
The nucleus of the cell is centrally important to an organism. It serves to store and organize genetic information, the atomic blueprint for the organism, while separating and protecting this very important information from the host of other cellular components. While the nucleus requires this protective isolation, it also needs to communicate with the rest of the cell, exchanging proteins and RNA, for a variety of nuclear and cytoplasmic processes which act in concert. The nuclear pore complex (NPC), perhaps the largest protein complex in the cell, is responsible for the protected exchange of components between the nucleus and cytoplasm and for preventing the transport of material not destined to cross the nuclear envelope. The large size of the NPC makes it difficult to study experimentally. Computational efforts can go a long way toward revealing properties of the NPC which are inaccessible by experiments. Recent molecular dynamics simulations have revealed interactions between the transport receptor importin-β and key nuclear pore proteins, bringing forth a better understanding of the selectivity of entry and exit from the nucleus.
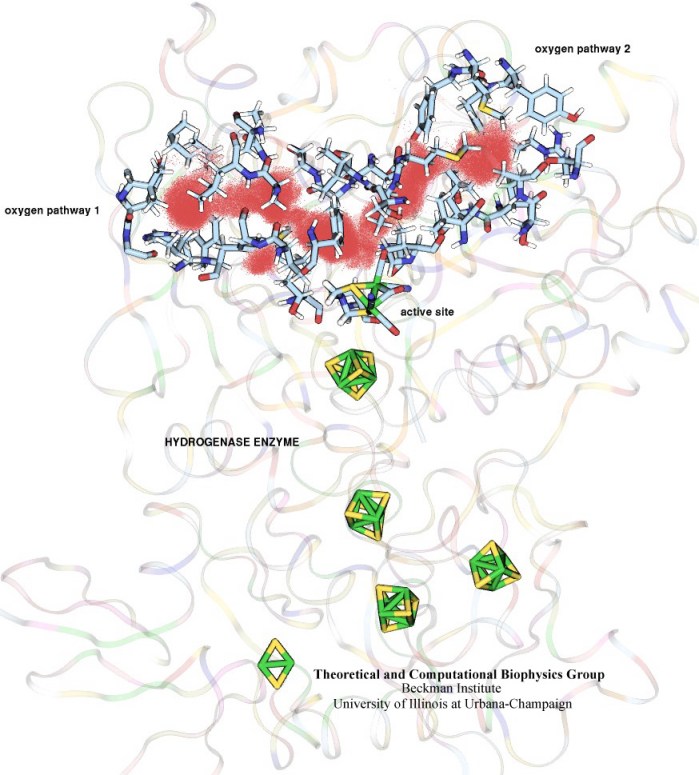
image size:
102.1KB
made with VMD
In an optimistic future, cars and appliances will be powered by renewable energy produced by burning hydrogen gas, with water being the only waste product. To supply this hydrogen gas, scientists are turning their attention to an enzyme called hydrogenase that is found in certain microorganisms, which produce hydrogen gas from sunlight and water. This enzyme, however, is sensitive to oxygen gas, which irreversibly deactivates its hydrogen-producing active site. Understanding how oxygen reaches the active site will provide insight into how hydrogenase's oxygen tolerance can be increased through protein engineering, and in turn make hydrogenase an economical source of hydrogen fuel. In a recent paper (also described in this webpage), the programs NAMD and VMD are used to analyze the gas diffusion process inside hydrogenase, and how it correlates with the protein's internal fluctuations, thereby creating a map of the oxygen pathways. The calculations revealed two distinct pathways for oxygen to reach the active site. Gases participate in physiological processes of many organisms and the new computational strategy developed promises to image gas diffusion pathways for many relevant proteins. In fact, the researchers are currently inspecting hundreds of proteins for their ability to internally transport gas molecules.
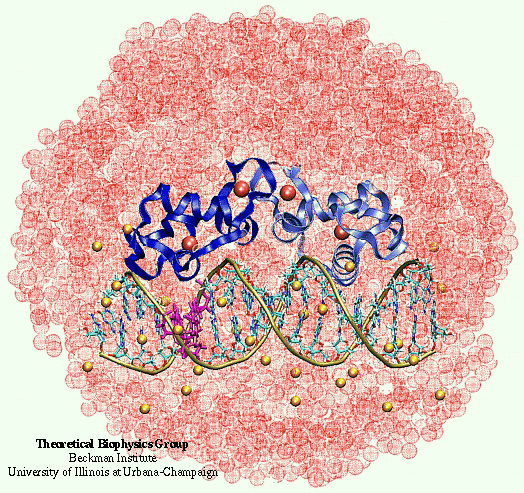
image size:
184.0KB
made with VMD
Nuclear hormone receptors are cellular regulators which activate the transcription of specific genes in response to the binding of nuclear hormones. We studied the specificity of DNA recognition by the estrogen receptor protein. The role of water molecules at the protein-DNA interface and changes in the DNA structure between specific and non-specific binding were monitored and analyzed.

image size:
18.7KB
made with VMD
Through modelling and quantum chemical studies, the group is supporting the design of novel proteins in collaboration with M. Sarikaya, U. Washington, Seattle. It is hypothesized that electrostatic interactions between the polar residues of this genetically engineered polypeptide and the gold surface allow stronger adsorbtion onto the {111} surface than to other Au crystal faces, thus influencing the crystalization of gold in the presence of this polypeptide.
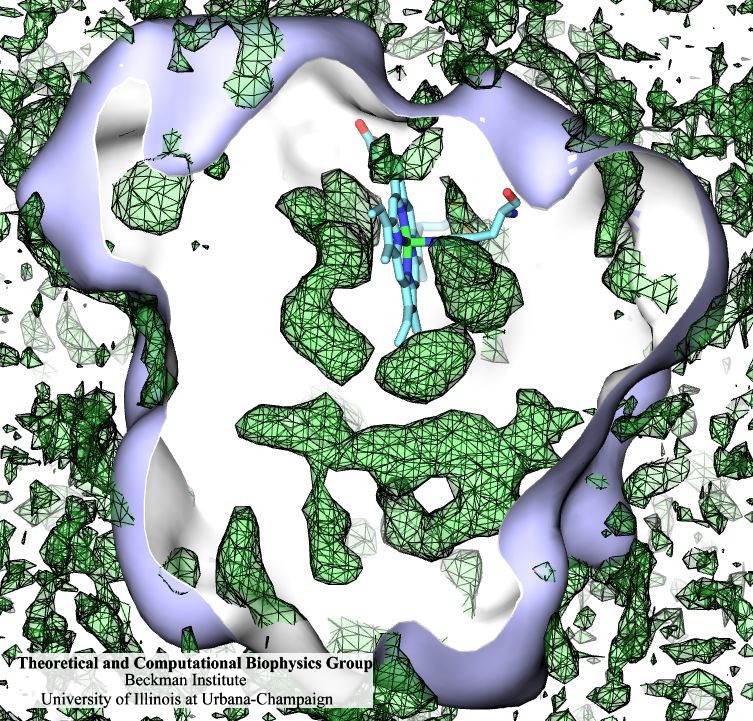
Many proteins interact with gas molecules such as oxygen to perform their functions. In most cases, the gas molecules must reach sites buried deep inside the proteins that bind the molecules, with no obvious way in. Understanding how, for example, oxygen enters the protein, and mapping out which pathways it takes has been a long-standing challenge. As reported recently, computational biologists, inspired by previous work on the hydrogenase enzyme (see the September 2005 highlight), have developed a method, called implicit ligand sampling, that maps the pathways taken by gas molecules inside proteins. The mapping is determined by monitoring fluctuations of the protein, surprisingly, in the absence of the gas molecules. The mapping method is available in the most recent version of the program VMD used for structure and sequence analysis of proteins. The researchers applied the method to myoglobin, an oxygen-storing protein present in muscle cells, and determined detailed three-dimensional maps of oxygen and carbon monoxide pathways inside the protein (for more information see our web page). While some details of these pathways were already known from experiment, the implicit ligand maps revealed a large number of new pathways and suggest that oxygen enters myoglobin using many different entrance doors.

image size:
118.5KB
made with VMD
movie (YouTube)
Antiretroviral therapies against human immunodefficiency virus type 1 (HIV-1) have proven to be extremely effective to control viral load and infectivity. However, the virus is increasingly faster acquiring new resistance to antiretroviral drugs used in current treatments. Viral particles of HIV-1 contain conical cores (or ``cones'') formed by a protein shell composed of the viral capsid protein (CA). The capsid protein (CA) plays critical roles in both late and early stages of the infection process and is widely viewed as an important unexploited therapeutic target that could offer the best hope of generating drugs that are active against all HIV-1 variants. The study of the nature of HIV-1 disassembly has been particularly difficult due to the vulnerability of the capsid assembly to experimental manipulation. Although early work suggested that disassembly occurs immediately following viral entry in the cell, thus attributing a trivial role for the capsid in infected cells, recent data suggest that uncoating occurs several hours later and that the capsid has a pivotal role in several stages of the infection process, namely for transport towards the nucleus, reverse transcription and nuclear import. These findings suggest that the viral capsid interacts with the cytoskeleton and other cytoplasmic components of the host cell during its transport to the nucleus and that its stability is crucial for the success of the virus.
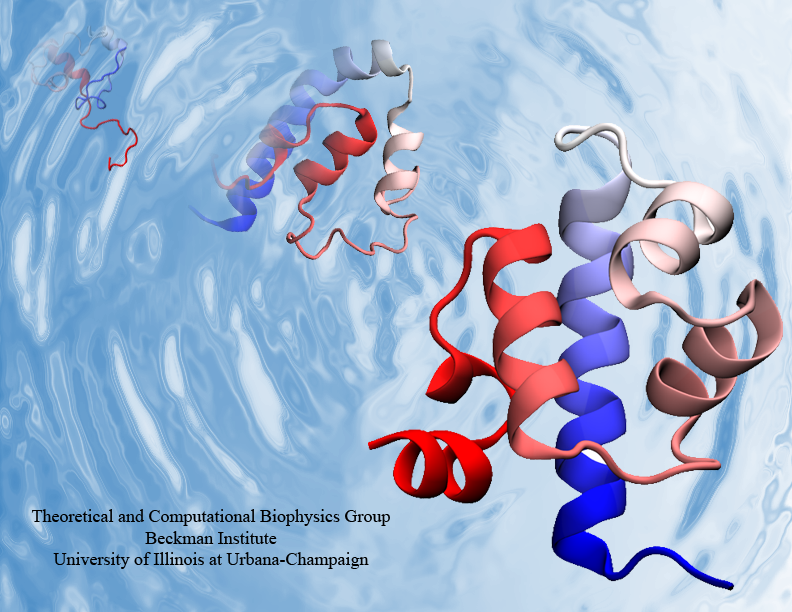
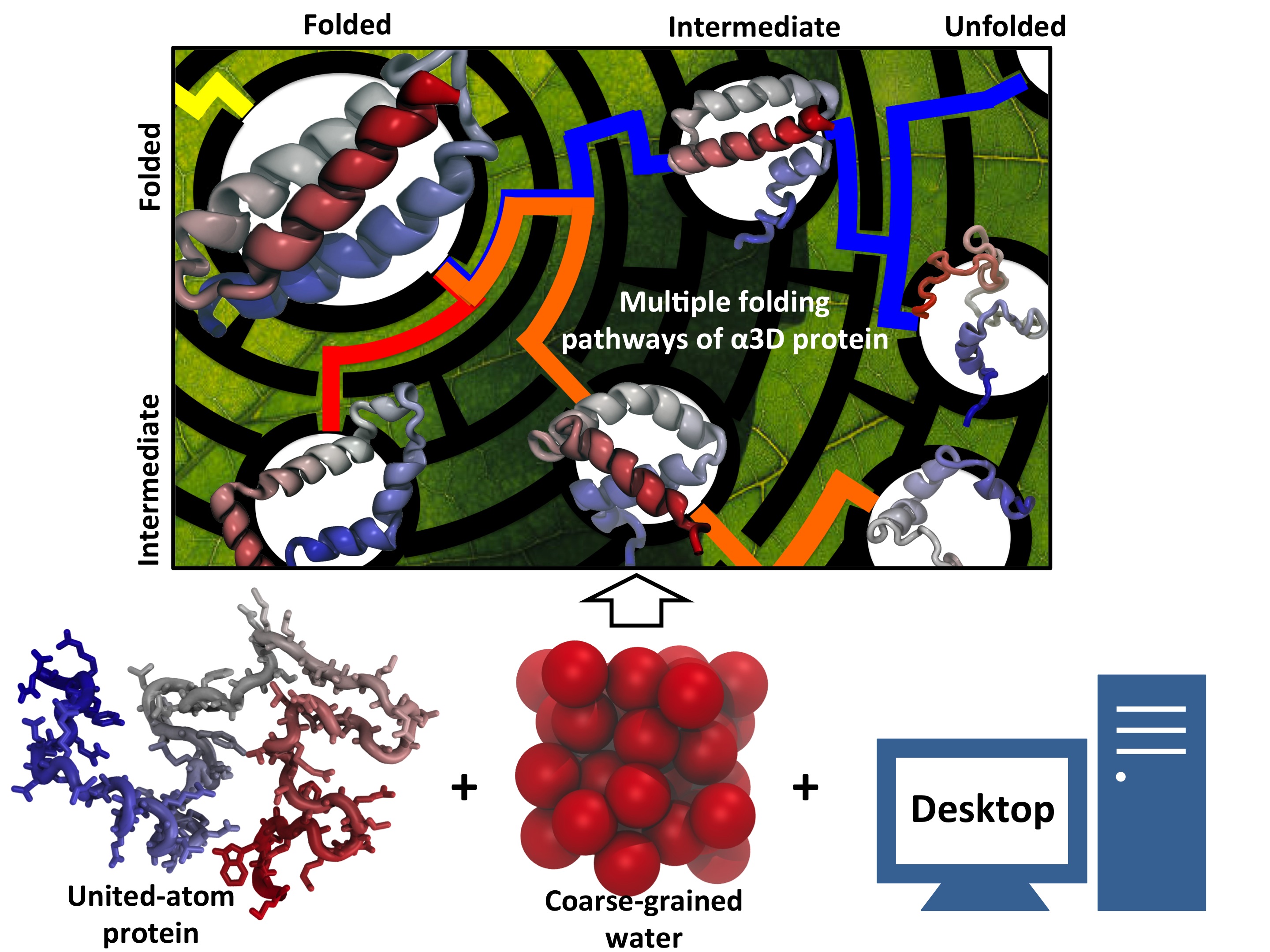
Proteins, made up of specified sequences of amino acid building blocks, are the workhorses of biological systems, performing most of the tasks necessary to maintain life. One of the greatest challenges in molecular biology today is that of determining how the sequence of a protein -- the exact ordering of amino acids it is composed of -- specifies its structure and function. Protein folding mechanisms have been extensively studied over the past few decades through both experimental and computational means, although the long timescales required for folding processes have meant that simulation of complete folding trajectories in explicit solvent were not possible until very recently. Instead, protein folding simulations have generally made use of coarse models, implicit solvent, or the use of very large ensembles of shorter trajectories to obtain information on the physical folding pathway. Recent advances, however, have made combined experimental and computaitonal studies of protein folding possible through the development and proteins that fold on the microsecond and even sub-microsecond timescale, and through advances in molecular dynamics (MD) simulations allowing simulation of multiple microsecond folding trajectories within a few months on modern supercomputers.
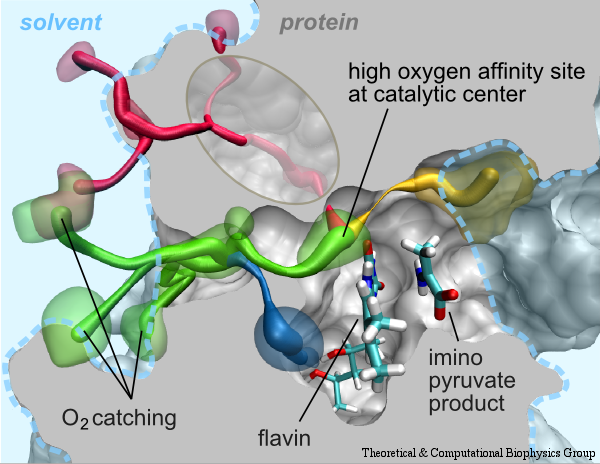
image size:
225.2KB
made with VMD
Molecular oxygen participates in numerous cellular processes but in most cases little is known about the mechanisms how oxygen reaches the reaction sites of the related proteins. However, in recent years specific oxygen access channels have been revealed for a number enzymes such as lipoxygenase, copper amine oxidase or glucose oxidase. In guiding oxygen to the catalytic site and by providing specific environmments for oxygen and the according reaction products these channels play significant roles in controlling O2 reactivity. There are many more promising candidates for oxygen access channels, especially among monooxygenases and oxidases from the flavoprotein family. In these enzymes, where O2 (re)oxidizes the reduced flavin cofactor, key questions regarding the control of O2 reactivity with the reduced flavin cofactor are still open.
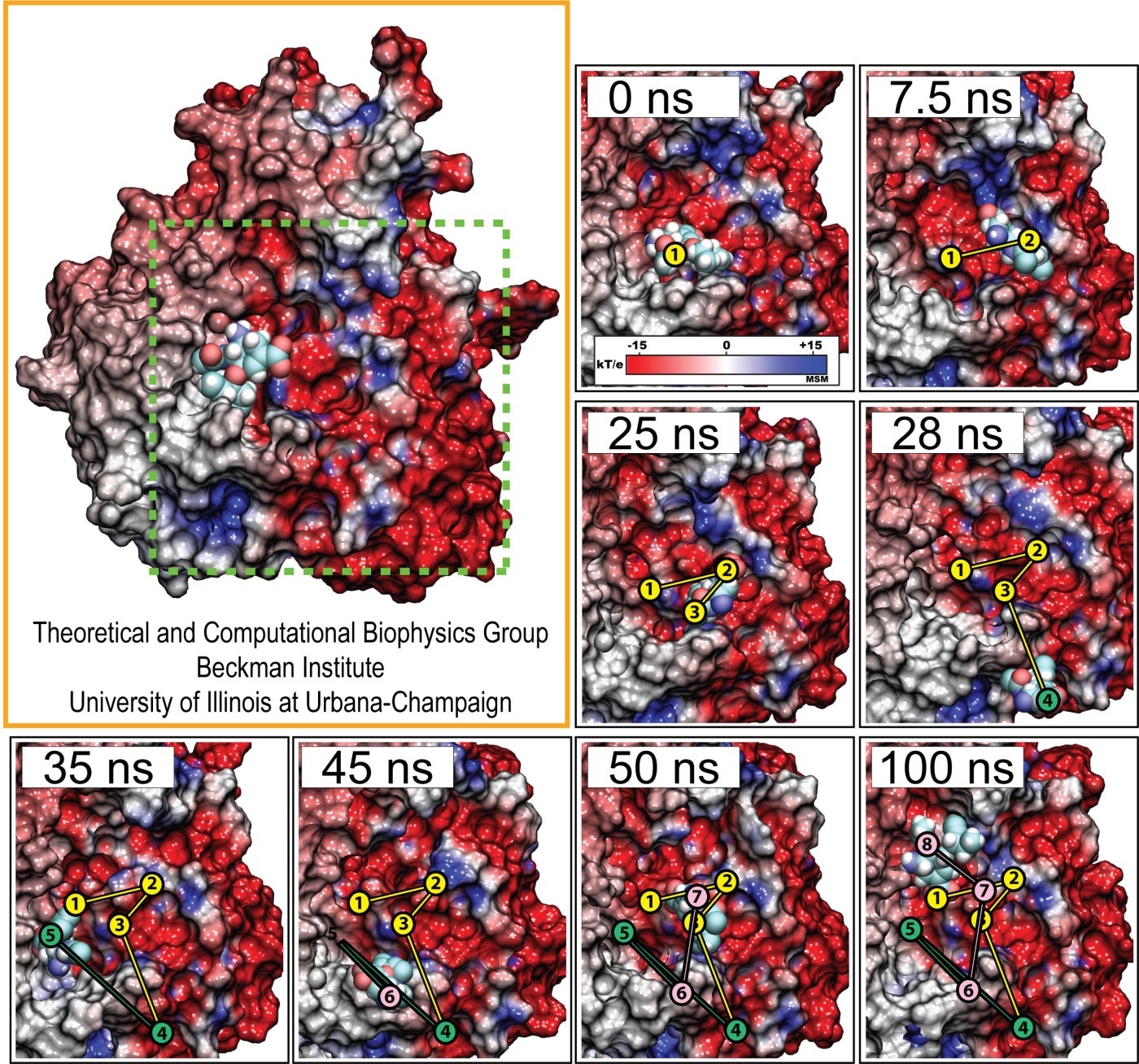
The 2009-2010 outbreak of the H1N1pdm "swine" influenza caught not only its victims by surprise, but also biologists who had assumed that the virulence (and ultimately, lethality) of human flu viruses were generally limited to the elderly and immunocompromised. Most fatal cases of H1N1pdm, however, involved young previously healthy people, raising the possiblity that H1N1pdm may eventually mutate into a strain capable of killing even healthy individuals, such as in the 1918 flu pandemic where an estimated 50 to 100 million people died (approximately 3% of the 1.6 billion world population at the time). Even more alarming however, was that while the intiial strains of H1N1pdm appeared to be susceptible to conventional antiviral therapy, the strain has mutated quickly to become drug-resistant to the front line antiviral drug, oseltamivir (Tamiflu). Oseltamivir normally functions by binding to the flu protein neuraminidase and preventing the virus from budding out of its infected host cell after replication. This alarming discovery, that the virus has gained an upper hand on medicine, has raised grave concerns regarding an effective defense against subsequent influenza outbreaks. The H1N1pdm "swine" influenza virus is closely related to the 2003 H5N1 "avian" influenza virus for which oseltamivir-resistance due to two individual point mutations, H274Y and N294S, is well known. However, the mechanism behind why these mutations actually inhibit successful binding of oseltamivir to neuraminidase is not well understood. Furthermore the mutations are actually located some distance from the drug-protein binding site, suggesting that the mutations may disrupt the actual drug binding process rather than just forming an inhospitable environment for drug-protein endpoint interactions.

image size:
77.5KB
made with VMD
Influenza viral infections affect all populations of the world and represent a leading cause of mortality in elderly and immunecompromised populations. The 2009 A/H1N1 pandemic, now believed to have caused as many as ten times more deaths than originally estimated, clearly illustrated how drug resistant mutants can impact a population before a vaccine is available. The development of potent and effective antiviral drugs, therefore, is of paramount importance for stemming future epidemics. The influenza virus capsid contains two glycoproteins, namely hemagglutinin and neuraminidase. Neuraminidase in particular binds with sialic acid on respiratory tract epithelial cells, allowing attachment of the virus and the subsequent release of viral replicants. This sialic acid binding site is a viable target for neuraminidase inhibitors such as Tamiflu (oseltamivir) and Relenza (zanamivir); however, the emergence of drug resistant mutations (H274Y, N294S, and Y252H) to the A/H1N1 strain as well as the more common H5N1 strain has significantly limited the effectiveness of Tamiflu in particular, inducing a drug resistance anywhere from 81- to 256-fold over the wild type strains.
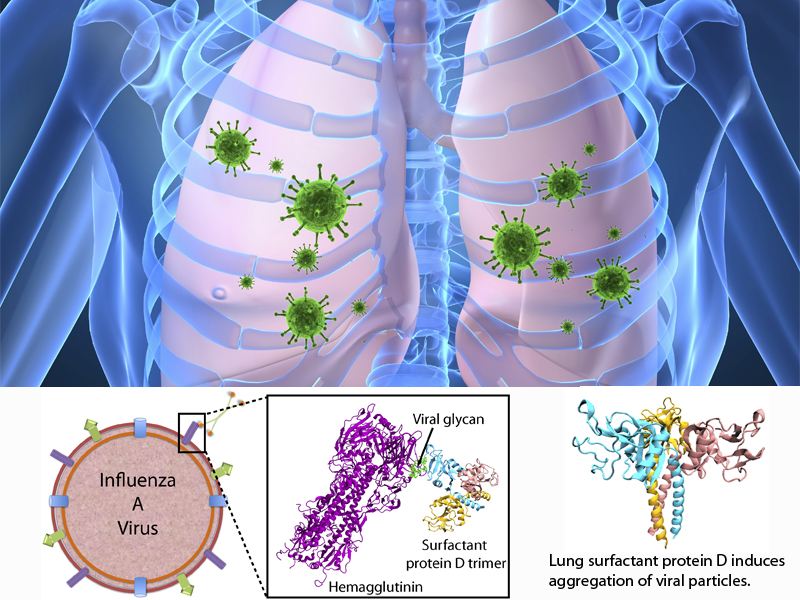
image size:
76.8KB
made with VMD
Influenza is a highly contagious respiratory disease, estimated to cause 250,000 to 500,000 deaths worldwide each year. Before Influenza A virus (IAV) infection of the host is established, the virus encounters various lines of innate defenses, including an important class of mammalian innate immune proteins, namely proteins called collectins. Lung collectins are involved in the early pulmonary response to limit infection and spread of IAVs and other pathogens in the airways. By now, it is well established that among these collectins, surfactant protein D (SP-D) appears particularly important in the context of IAV. SP-D-mediated protection is primarily established by reducing the number of infectious particles via aggregation of viral particles, which prevents attachment of virus to the host respiratory epithelium and induces phagocytic responses resulting in enhanced viral clearance.
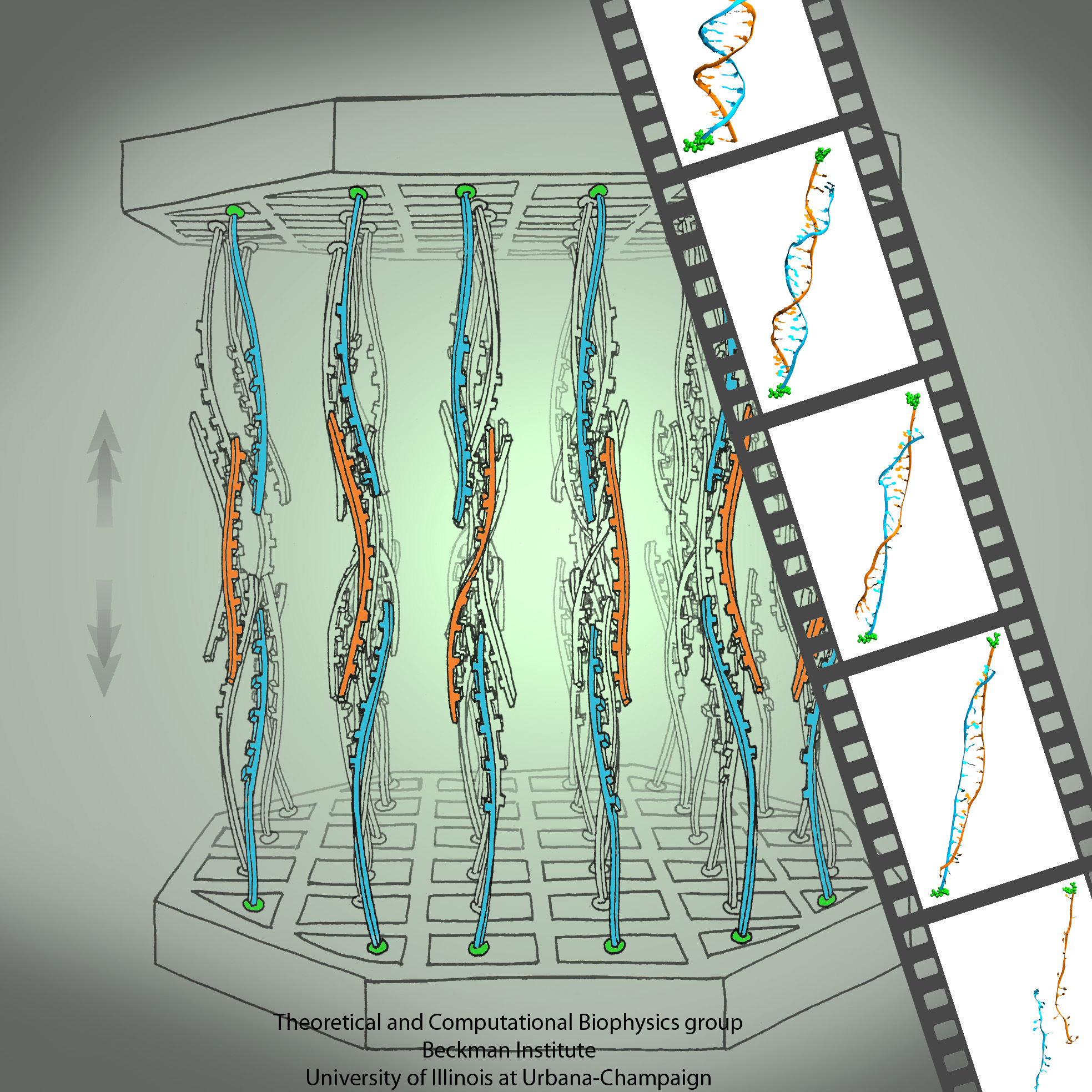
Methylation of cytosine is a covalent modification of DNA, in which hydrogen H5 of cytosine is replaced by a methyl group. In mammals, 60% - 90% of all CpGs are methylated. Methylation adds information not encoded in the DNA sequence, but it does not interfere with the Watson-Crick pairing of DNA - the methyl group is positioned in the major groove of the DNA. The pattern of methylation controls protein binding to target sites on DNA, affecting changes in gene expression and in chromatin organization, often silencing genes, which physiologically orchestrates processes like differentiation, and pathologically leads to cancer.
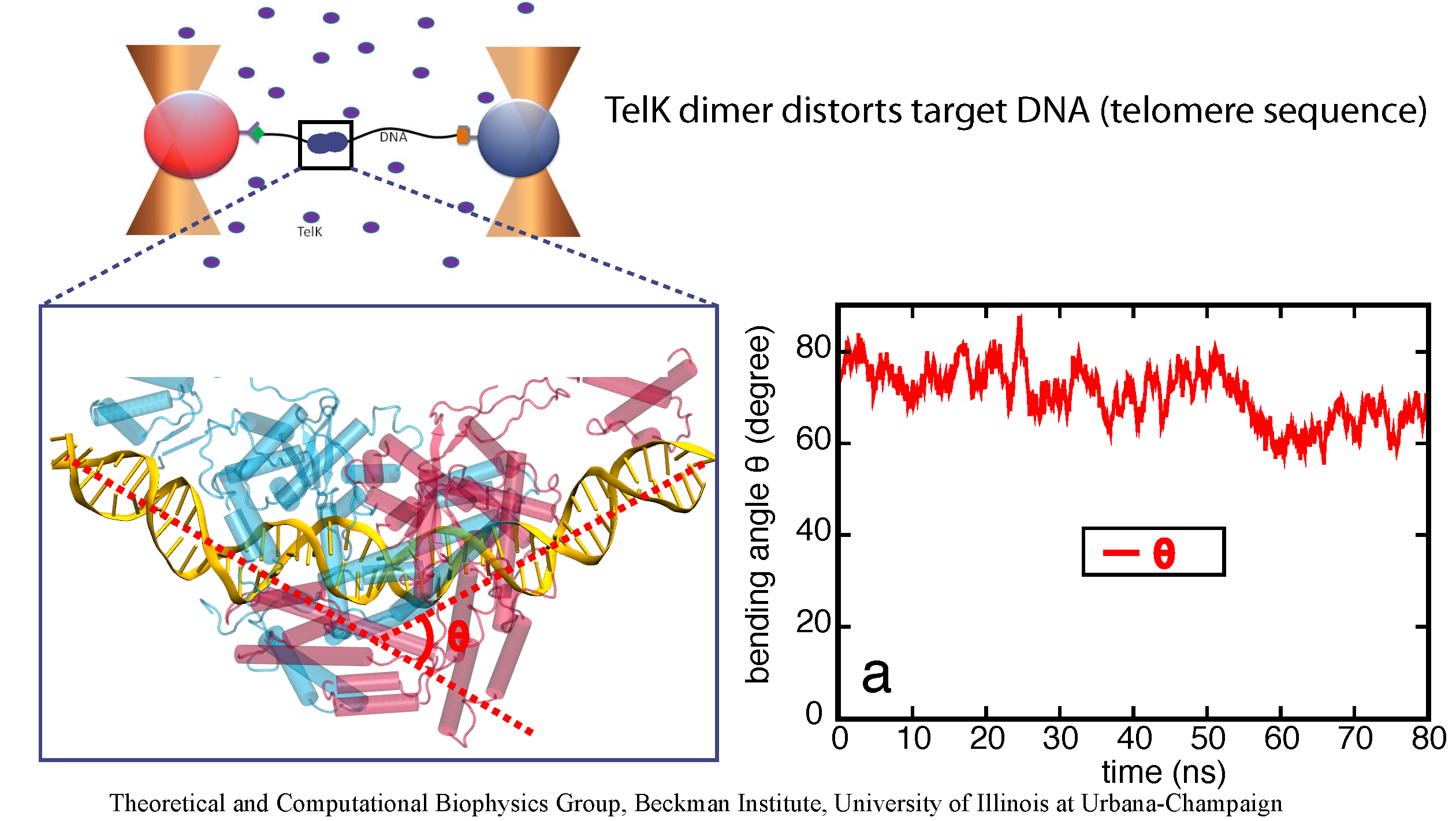
image size:
651.0KB
made with VMD
DNA, a long linear molecule, is the carrier of genetic information. In the cell, each DNA molecule is packaged in a structure called chromosome. The ends of linear chromosomes are capped by structures known as telomeres to prevent fusion with neighboring chromosomes. Telomeres are maintained by an enzyme called telomerase during DNA replication. In order to do so, telomerase has to find the telomere region on DNA quickly and precisely. One telomerase is the protelomerase TelK, which binds to the ends of DNA, cleaves DNA strands and refolds cleaved DNA ends into hairpin telomeres in linear chromosomes of prokaryotes and viruses. Previous studies have shown that TelK is only active as a dimer. Researchers investigated the target-search mechanism of protelomerase TelK through single-molecule experiments and molecular dynamics simulation. It was revealed that as a monomer, TelK undergoes one-dimensional diffusion along non-specific DNA (without telomere sequence), and is able to bind to the target site preferentially. There, the target-immobilized monomer waits for a second binding partner to form an active protein complex. More on our TelK website.
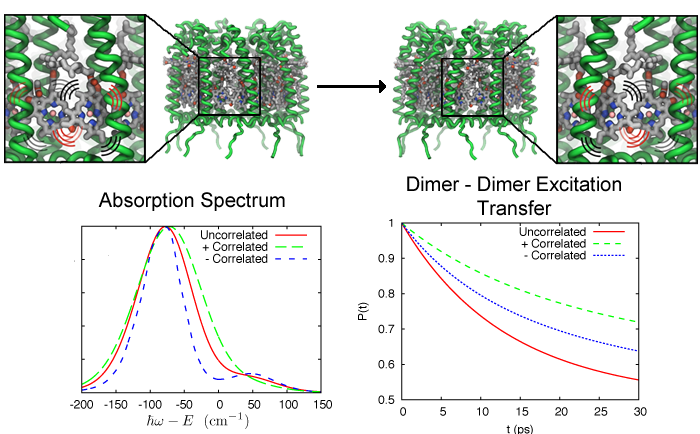
image size:
231.6KB
made with VMD
Excitation transfer between pigment molecules, such as bacteriochorophylls (BChls), and between pigment-protein light harvesting complexes, such as light harvesting complex 2 (LH2), has been investigated for many years using many different theoretical descriptions. Typically these descriptions include a priori assumptions about the dynamics of the system. Such assumptions are often made due to incomplete knowledge to make the system numerically tractable so that further insight can be gained. In the theoretical models of excitation transfer, it is often assumed that one parameter is much larger than another, allowing the system to be treated perturbatively. These assumptions, however, should be physically reasonable and should be tested if possible.
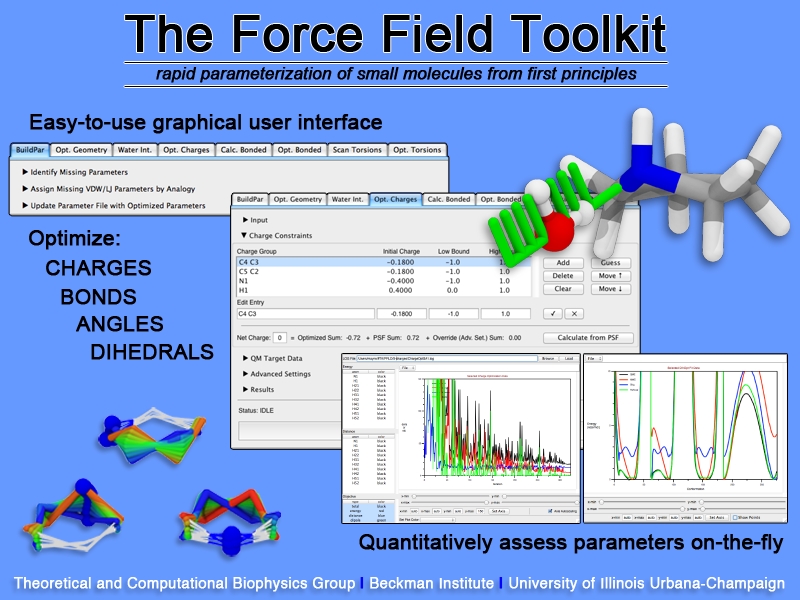
image size:
307.6KB
Structural biologists are increasingly turning to simulation methods to investigate the connections between molecular structure and biological function. Classical molecular dynamics (MD) simulations, such as those performed by the simulation software NAMD, rely on potential energy functions requiring parameters to describe atomic interactions within the molecular system. While these parameters are available for the most commonly simulated biopolymers (e.g., proteins, nucleic acids, carbohydrates), many small molecules and other chemical species lack adequate descriptions. The complexity of developing these parameters severly restricts the application of MD technologies across many fields, including most notably drug discovery. Researchers have developed software, the Force Field Toolkit (ffTK), that greatly reduces these limitations by facilitating the development of parameters directly from first principles. ffTK, distributed as a plugin for the molecular modeling softare VMD, addresses both theoretical and practical aspects of parameterization by automating tedious and error-prone steps, performing multidimensional optimizations, and providing quantitative assessment of parameter performance--all from within an easy-to-use graphical user interface. Additional information on ffTK, including documentation and screencast tutorials, can be found here.
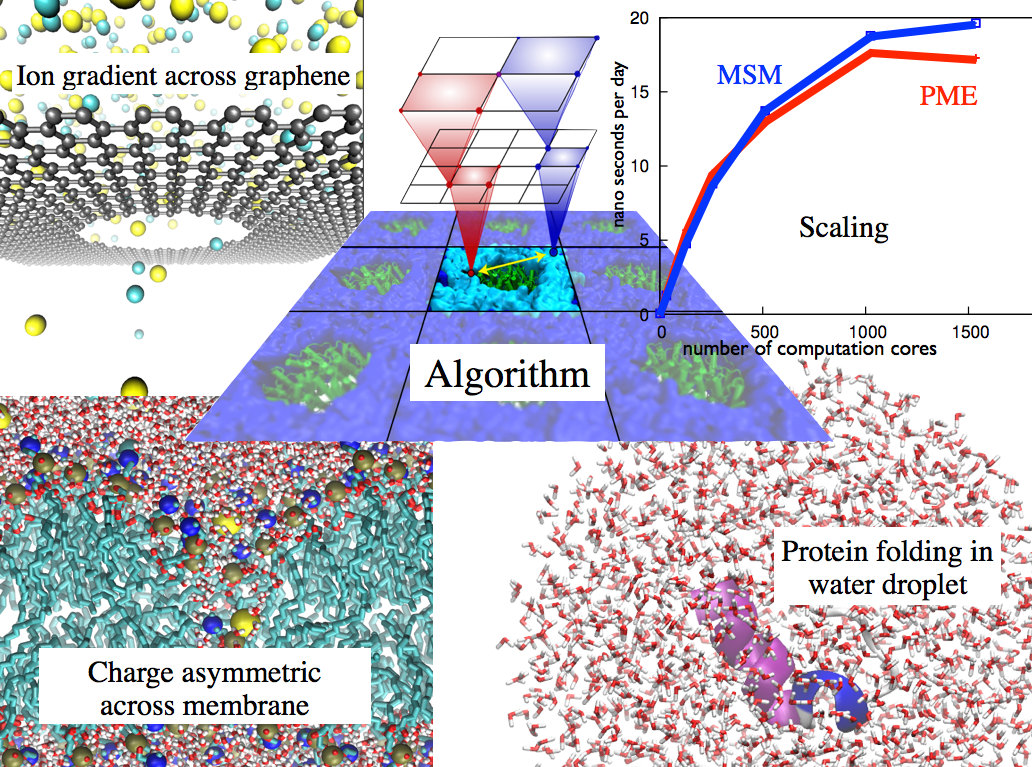
image size: 351.4KB



{kind=link}
{kind=link}