Imaging gas migration pathways inside proteins
Oxygen-breathing proteins
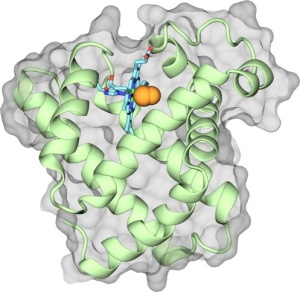
Fig 1. Representation of myoglobin, highlighting its prosthetic group, the heme (in colored licorice), which is holding on to a bound CO molecule (orange).
Charting a Map
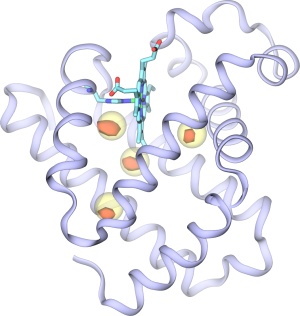
Fig. 2. Predicted xenon binding sites form implicit ligand sampling (red) along with experimentally measured locations of xenon atoms (yellow) in myoglobin.
Lost in myoglobin
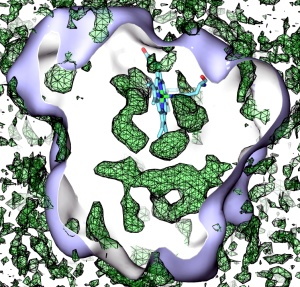
Fig. 3. A 3-D map of myoglobin showing the areas accessible to O2, in green. The empty space not enclosed by these surfaces corresponds to areas that are not easily accessible to O2. Also see movie.
We applied the implicit ligand sampling method to the oxygen-binding protein myoglobin. This protein has been studied experimentally for its gas migration properties extensively over the past decades. The implicit ligand sampling was first used to create a map for xenon pathways. Xenon is often used by experimentalists to find favorable sites for gas molecules inside proteins. Due to its large size, xenon gets easily trapped in proteins at specific locations which can then be identified using x-ray crystallography because of the high electron density of Xe atoms. Fig. 2 shows the locations of the xenon binding sites in myoglobin measured from x-ray crystallography, along with those predicted from molecular dynamics simulations; in the latter case the binding sites were identified as those places in the implicit ligand maps found to be energetically most favorable for Xe. Both methods, x-ray crystallography and implicit ligand sampling, suggest the Xe binding sites and predict similar occupancies for Xe.
We then created 3-D implicit ligand maps for O2, CO and NO in
myoglobin. Fig. 3 shows surfaces of equal free energy from the full
O2 map (also see accompanying movie). In the movie, one can see the well-known
distal exit which leads right from the heme binding site directly
to the external solution. What is more surprising is the large number of
additional exits for O2, especially those far away from the
heme. While many of the cavities neighboring the heme were experimentally
known to host O2, we did not expect that myoglobin would be so
porous. The additional exits discovered by our computations are not easily
observed experimentally, and automated computational tools such as
implicit ligand sampling finally provide scientists with a way of easily
discovering and describing hard-to-find gas pathways in gas-binding
proteins.
O2 pathways are not conserved in globins
Now that we have a means of computing oxygen pathways inside proteins, we turn our attention to establishing what these pathways look like, not just for myoglobin, but for the globin superfamily. To do this, we picked twelve monomeric globins of known structure and computed PMF maps of their internal oxygen pathways. What we saw was unexpected (Cohen et al., 2007). As a whole, the globin family exhibited no conservancy of the pathways at all between different globins, despite the fact that every globin examined shared an almost-identical protein fold. An alignment of the different globin structures highlighting the conserved globin fold, along with the computed oxygen pathway networks inside selected globin, highlighting the lack of pathway conservancy, is shown in Fig 4.
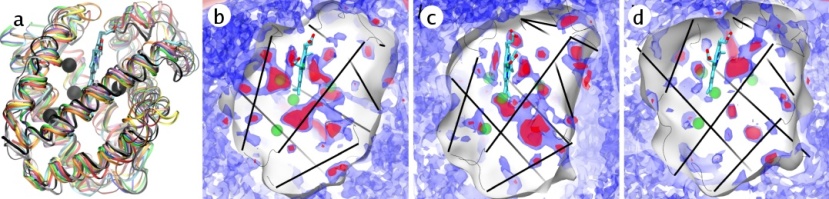
Fig 4. (a) The alignment of the structures of twelve monomeric globins shows that the globin fold is well conserved. Despite this, there is wide variation in the internal networks of oxygen cavities and pathways between globin species. Shown, are the free energy isosurfaces for O2 (red is very favorable, blue is merely favorable, and white is inaccessible) for three of the globins that we studied: (b) roundworm hemoglobin (c) trematode hemoglobin and (d) soy leghemoglobin. The heme groups are shown as licorice, and the globin alpha-helices are drawn as black lines.
These results are important because they demonstrate that not all global properties of proteins can be assumed to be consereved for similar protein folds and structures. What is special about the case of oxygen pathways is they are not determined by the protein's "static structure" per se, but rather by the local dynamics of the protein's residues. In fact, when one examines the propensity of individual residue types to lie near or far away from pathways, one finds that large hydrophobic residues tend to promote the formation of O2 pathways. This appears paradoxal, because from a "static structure" point of view, bulky residues would have a tendency to fill up voids in the protein. However, when one looks at the dynamic picture, bulky hydrophobic residues interact weakly with their neighbors and are very mobile: this is the perfect combination for forming oxygen-favorable regions inside proteins.
O2 uptake in copper amine oxidases
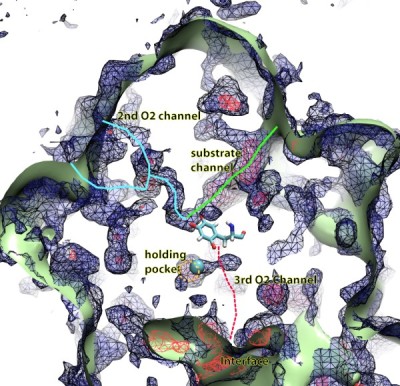
Fig. 5. Map of the oxygen pathways in the copper amine oxidase enzyme from Hansenula polymorpha
Another group of proteins that uses molecular oxygen are those from the copper amine oxidase (CAO) family. CAOs manufacture various organic compounds by reacting the O2 with an amine group-containing substrate. Researchers have been wanting to know how the CAO's various substrates reach its active site before the enzymatic reaction occurs. But while all CAOs exhibit a clear channel for the amine substrate to reach the copper active site, the are no obvious oxygen entry pathways.
To answer this question, we have computed and mapped the probability of finding O2 everywhere inside four CAOs from different species using implicit ligand sampling (Johnson et al., 2007). From these detailed maps, one of which is shown in Fig. 5, we were able to accurately predict and identify the majority of all the experimentally determined xenon binding sites in these four CAOs (despite the fact that the actual locations of Xe binding sites were kept secret from the computing team by the experimental team). Just like for the case of myoglobin, in addition to reproducing experimental results, our computed O2 probability maps reveal more than just the location of the xenon binding sites; it revealed the entire network of O2-accessible pathways inside the proteins.
Across the four studied CAOs, many different O2 pathways were
found. Instead of a unique and precise O2 channel leading from
the external solution into the copper active site, as was previously
assumed, O2 can enter CAOs following many pre-determined
pathways, as if the CAOs were sponges. This result explains the fact that
many mutation along previously hypothesized O2 "channel"
locations had little effect on the CAOs' reaction rates. Furthermore, the
O2 pathway maps revealed that what was previously a main
candidate pathway for O2 entry -- a short water file leading to
the active site -- was in fact not very permeable to O2 at all.
Had this been the case, O2 would be able to react with the
copper active site in advance of when this is desirable for the CAO's
proper function, and would have slowed down CAO's enzymatic rate. Instead,
we propose that the short water file really serves as an exit channel for
the polar peroxide (H2O2) product arising from the
enzymatic reaction.


