Molecular Basis of Drug Resistance in A/H1N1 Influenza Virus

Overview
Influenza viral infections affect all populations of the world and represent a leading cause of mortality in elderly and immunecompromised populations. The 2009 A/H1N1 pandemic, now believed to have caused as many as ten times more deaths than originally estimated, clearly illustrated how drug resistant mutants can impact a population before a vaccine is available. The development of potent and effective antiviral drugs, therefore, is of paramount importance for stemming future epidemics.
The influenza virus capsid contains two glycoproteins, namely hemagglutinin and neuraminidase. Neuraminidase in particular binds with sialic acid on respiratory tract epithelial cells, allowing attachment of the virus and the subsequent release of viral replicants. This sialic acid binding site is a viable target for neuraminidase inhibitors such as Tamiflu (oseltamivir) and Relenza (zanamivir); however, the emergence of drug resistant mutations (H274Y, N294S, and Y252H) to the A/H1N1 strain as well as the more common H5N1 strain has significantly limited the effectiveness of Tamiflu in particular, inducing a drug resistance anywhere from 81- to 256-fold over the wild type strains.
The recent elucidation of the crystal structure of A/H1N1 neuraminidase (PDB ID: 3NSS), provides a framework for capturing the mechanisms of drug binding aimed at this protein specifically. On the basis of this novel three-dimensional structure, we employed molecular dynamics simulations to analyze the binding of Tamiflu and Relenza to wild-type and mutant neuraminidases and to characterize the drug-resistance resulting from the H274Y, N294S, and Y252H mutations. At the energetic level, we performed a thermodynamic analysis of the association process using free-energy perturbation (FEP) calculations, with the objective of measuring the relative binding affinity of A/H1N1 neuraminidase mutants to selective inhibitors of the A/H1N1 virus.
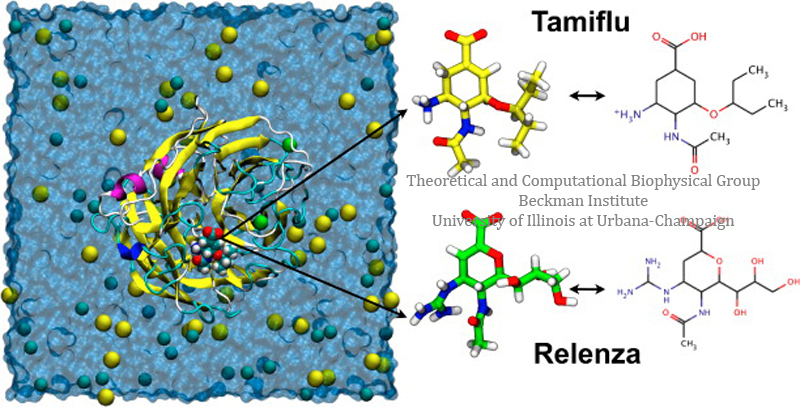
Overview: Antiviral drugs bound to A/H1N1 influenza neuraminidase.
Results
Wild type complexes – conserved drug-protein interactions and calculation of binding affinities
To evaluate the specific interactions of Tamiflu and Relenza with the wild-type A/H1N1 neuraminidase, structural analyses of equilibrated complexes were performed. The results show that the hydrogen-bond network is well conserved for both drugs; the complex with Tamiflu exhibits seven hydrogen bonds on average, while that featuring Relenza is stabilized by ten hydrogen bonds. Specifically, both neuraminidase inhibitors share the same binding pocket,18,19 which includes hydrogen bonds to residues R117, E118, D150, E277, R292, and R371 of the neuraminidase; Relenza, however, forms additional hydrogen bonds with residues E227 and E276. Although the residues surrounding both drugs are highly conserved, the guanidinium and hydroxyl groups of Relenza allow a hydrogen-bond network to be formed, markedly more robust than that for Tamiflu.
It is well known that in addition to the well-conserved hydrogen-bond network, the electrostatic environment within the active site contributes to drug-binding stability. The three-dimensional electrostatic potential of the equilibrated complexes was calculated and averaged for each trajectory. In the wild-type neuraminidase:Tamiflu complex, the residues around the drug are oriented in such a way as to form an electronegative surface within the binding pocket, while the entrance to the active site has some electropositive and electronegative areas. On the other hand, the binding pocket residues surrounding Relenza show less electronegative area than for Tamiflu and the entrance to the active site tends to be electronegative. Therefore, while the network of hydrogen bonds is larger for Relenza, the electrostatic potential is more favorable for Tamiflu, leading to similar affinities and, therefore, to similar inhibitory effects. The residues that contribute predominantly to the electrostatic potential of the binding pocket for both drugs are R117, E118, L133, D150, R151, W178, S179, I222, R224, E227, S246, E276, E277, R292, N294, R371, and Y406.
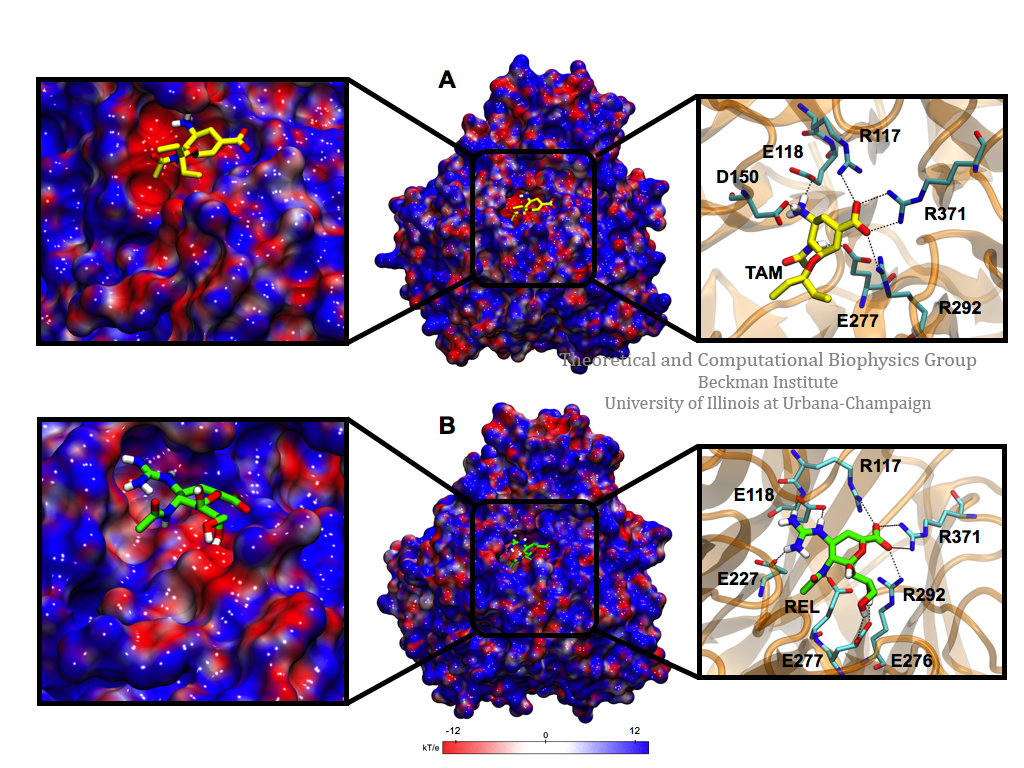
Figure 1. Electrostatic potential surface and hydrogen-bond network of the binding pocket of A/H1N1 neuraminidase in interaction with Tamiflu and Relenza. Complete map (center) and a close-up (left) of the binding pocket with Tamiflu (A, in yellow) and Relenza (B, in green) bound to A/H1N1 neuraminidase. Hydrogen bonds between residues in the binding pocket and each drug are also shown (right). These hydrogen bonds are generally the same between the drugs, although Relenza shows additional interactions with E227 and E276. The internal cavity exhibits a negative electrostatic potential (in red) and the region around the binding site possesses a positive electrostatic potential (in blue) that interacts with the −NH2 and −COO– moieties of both drugs. The electrostatic potential surfaces were generated using the APBS software interfaced through VMD.
The relative binding affinity of Tamiflu and Relenza in complex with the wild-type and mutant A/H1N1 neuraminidases were separately calculated using free-energy perturbation (FEP). Both mutants show an unfavorable energy (shown as Bennett acceptance ratio, or BAR, estimates in Table 1) in the presence of Tamiflu, suggesting a higher resistance to this drug and, hence, a better inhibitory capacity of Relenza in comparison with Tamiflu. On the other hand, the Y252H mutant leads to a BAR net free-energy change suggesting that Tamiflu remains an effective inhibitor for the Y252H mutant. Overall, the present set of results shows an excellent agreement (∼90%) with the experimental estimates (see Table 1), confirming the ability of the computational approach used here to predict relative binding affinities of drugs against a specific protein.
Table 1. Bennett acceptance ratio (BAR) estimates were obtained by combining the forward and backward transformations
Drug resistance mutations induce local electrostatic changes to the active site
Next, a structural analysis of the mutant:drug complexes was performed to characterize the molecular interactions lost in the case of drug resistance. Hydrogen-bond and electrostatic-potential analyses for each complex in the initial state (λ = 0) and the final state (λ= 1) of the FEP calculations are shown in Figure 2. In comparison with the wild-type neuraminidase, the H274Y:Tamiflu complex conserves the hydrogen-bond network, albeit the local electrostatic surface potential differs, revealing a binding pocket less electronegative relative to the wild-type. At the same time, the region that includes the mutated (H274Y) residue is observed to be more electropositive, suggesting that the affinity of the drug is altered in part by a change in the electrostatic potential due to a spatial reorganization of residues within the neighborhood of H274Y. In the case of Relenza, the electrostatic potential, compared to wild-type complex, becomes slightly more negative in the binding pocket and is rather more positive in the region in contact with the hydroxyl groups of the drug, which likely explains in part the affinity of Relenza versus Tamiflu in presence of this mutant. Similarly, the N294S mutant interacting with either drug exhibits a similar hydrogen-bond network, although for Tamiflu the hydrogen bond with E277 is replaced by a new hydrogen bond with R151. For Relenza the interactions with R117, E276, and R371 are lost, while new hydrogen bonds with D150, R151, and S246 appear. It is likely, then, that the spatial reorganization of these residues contribute to the change of electrostatic potential within the binding pocket of the N294S:Tamiflu complex in comparison to the wild-type. Meanwhile, the electrostatic potential surface of the N294S:Relenza complex becomes more negative in this region, and as in the previous mutant, a new electropositive area is observed in the vicinity of hydroxyl groups of the drug. Therefore, although the H274Y and N294S mutations do not substantially alter the number of hydrogen bonds, they appear to cause reorganization of neighboring residues, leading to a significant change in the local electrostatic potential perceived by the drugs.

Figure 2. Images of the active site in the wild-type and mutant states of A/H1N1 neuraminidase in association with Tamiflu and Relenza. Average electrostatic potential surface and hydrogen-bond network for Tamiflu and Relenza bound to A/H1N1 neuraminidase for the wild type and mutants (H274Y, N294S, and Y252H). The values λ = 0 and 1 indicate that the wild-type and mutants serve as the reference and target states, respectively, for the FEP calculations. Despite the fact that the mutations do not involve residues in direct contact with the drugs, they cause significant changes in the environment that makes direct contact with Tamiflu and Relenza.
Just as in the case of the mutants described above, the Y252H mutant exhibits for both drugs an electronegative binding pocket; however, the potential at the entrance of the active site shows a more negative potential for Tamiflu as compared to Relenza. Moreover, this mutant, in contrast with H274Y and N294S, shows a different side-chain orientation for residue H252 for Tamiflu or Relenza, which may explain the drastic change in the electrostatic potential. In addition, these structural analyses are consistent with the relative binding free energy calculated by FEP, that is, the mutations H274Y and N294S alter the electric potential of the binding pocket in a way that matches well with the charge distribution of Relenza, but not of Tamiflu. Specifically, the region of negative electrostatic potential of the binding pocket of both mutants coincides with the positively charged guanidinium group of Relenza, while the negatively charged hydroxyl groups of the drug coincide with the positive potential that form the residues surrounding them. In contrast, the remarkable negative binding pocket of the Y252H:Tamiflu complex yields a more favorable electrostatic interaction for Tamiflu than for Relenza, and Tamiflu, therefore, remains effective for the Y252H mutant, in agreement with the aforementioned computed relative binding free-energy.
The infiltration of water into the active site alters drug binding affinity of mutants
One well-known phenomenon that can substantially alter the electrostatic interaction of a drug and a protein is the infiltration of water molecules, which induces a reorganization of the residues that surround the active site and, therefore, entails a spatial redistribution of the charges. To analyze this infiltration, we turned our attention to examining the relative hydration of the drug binding pockets and the entrance to the active site (in the region of the mutants), evaluating the number of water molecules and the solvent accessible surface area (SASA) for each wild-type and mutated system. Our analyses showed that a larger number of water molecules were present in the active site of the mutants compared to that of the wild-type, suggesting that the drug affinity may depend on the hydrophobicity of the active site.
Analyses of the conformational changes caused by water infiltration further show that in both wild-type and mutant simulations, orientation of residues R117, E118, D150, R292, and R371 is well-conserved; however, displacement of the side chains of the other residues surrounding the active site are observed. Specifically, inspection of the H274Y mutant bound to Tamiflu reveals that the strong interaction of H274 and E276 is disrupted by the mutation H274Y, causing E276 to shift from its wild-type position, allowing water molecules to penetrate into the H274Y pocket (Figure 3A). In the case of Relenza, the mutated residue Y274 destabilizes the hydrogen bond between the amide group of the drug and the E276 residue, leading fewer water molecules to enter, compared to Tamiflu. These results lend support to the assertion that the change in electrostatic potential at the entrance of the binding site is caused by the entry of water molecules within the neighborhood of H274Y. The water molecules conspire to displace the amino acids located in this region, which implies a reorganization of the superficial charges surrounding the active site. For the N294S mutant interacting with Tamiflu and Relenza, similar changes are observed in the residues located around the drugs (Figure 3B). Finally, for the Y252H mutant complexed with Tamiflu, we do not observe a significant influx of water molecules, while when Relenza is present, the H252 residue is oriented toward the H274 residue, hence, permitting water molecules to interact with the E276 residue (Figure 3C). Consequently, the presence of water molecules in the vestibule of the binding pocket seems to contribute significantly to the conformational change of key amino acids surrounding the active site through a modification of the electrostatic environment felt by the drugs (see Table 2). Furthermore, the E276 residue for each simulated system appears to be a vital factor for the stability of Tamiflu and Relenza, since in our mutant systems it adopts conformations that disrupt the normal accommodation of the drugs in the active site.
Figure 3. Infiltration of water molecules in the entrance of the active site of mutants of A/H1N1 neuraminidase. Structural characterization of the binding site of the (A) H274Y, (B) N294S, and (C) Y252H mutants associated to Tamiflu (left) and Relenza (right). The entry of water molecules into the binding site of the drugs is directly related to the conformational state adopted by the E276 residue.
Summary
Our results reveal that the origin of the resistance to antiviral drugs for the H274Y, N294S, and Y252H mutants is caused by penetration of water molecules at the entrance of the binding site of neuraminidase. The change in the electrostatic potential arising from a spatial reorganization of the residues surrounding the binding site appears to be a major factor that governs drug selectivity toward different mutants. Relenza occupies greater physical space than Tamiflu, so that Relenza, located in the active site, prevents the ingress of water molecules, which, in the case of Tamiflu, penetrate deeply into the vestibule of the binding pocket and strongly alter the electrostatic signature of the residues pertaining to the neuraminidase active site. These results are expected to help guide the design of novel drugs with an increased antiviral efficacy.
Cover figure
This article was selected for the cover of JCIM Nov 2012. You may
view the cover here.Related publications
Molecular basis of drug resistance in A/H1N1 virus. Ariela Vergara-Jaque, Horacio Poblete, Eric H. Lee, Klaus Schulten, and Christophe Chipot. Journal of Chemical Information and Modeling, 52:2650-2656, 2012. (PMC:DNA/NIH)
Molecular dynamics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. Ly Le, Eric H. Lee, David J. Hardy, Thanh N. Truong, and Klaus Schulten. PLoS Computational Biology, 6:e1000939, 2010. (13 pages).
Molecular modeling of swine influenza A/H1N1, Spanish H1N1, and avian H5N1 flu N1 neuraminidases bound to Tamiflu and Relenza. Ly Le, Eric H. Lee, Klaus Schulten, and Thahn Truong. PLoS Currents: Influenza, 2009 Aug 27:RRN1015, 2010. (9 pages).
Related methods
More information about the FEP method can be found here at our Free Energy Pertubation Tutorial.Investigators
- Christophe Chipot (Centre National de la Recherche Scientifique (CNRS), Nancy, France)
- Ariela Vergara-Jaque (Universidad de Talca and Universidad Andrés Bello, Chile)
- Horacio Poblete (Universidad de Talca and Universidad Andrés Bello, Chile)
- Eric H. Lee
- Fernando Gronzalez-Nilo (Universidad de Talca and Universidad Andrés Bello, Chile)
- Klaus Schulten
Page created and maintained by Eric H. Lee.





{kind=link}