Molecular Mechanism of Tamiflu Resistance
Introduction
Ah choo...you have the flu!
The 2009-2010 outbreak of the H1N1pdm "swine" influenza caught not only its victims by surprise, but also biologists who had assumed that the virulence (and ultimately, lethality) of human flu viruses were generally limited to the elderly and immunocompromised. Most fatal cases of H1N1pdm, however, involved young previously healthy people, raising the possiblity that H1N1pdm may eventually mutate into a strain capable of killing even healthy individuals, such as in the 1918 flu pandemic where an estimated 50 to 100 million people died (approximately 3% of the 1.6 billion world population at the time). Even more alarming however, was that while the intiial strains of H1N1pdm appeared to be susceptible to conventional antiviral therapy, the strain has mutated quickly to become drug-resistant to the front line antiviral drug, oseltamivir (Tamiflu). Oseltamivir normally functions by binding to the flu protein neuraminidase and preventing the virus from budding out of its infected host cell after replication. This alarming discovery, that the virus has gained an upper hand on medicine, has raised grave concerns regarding an effective defense against subsequent influenza outbreaks.
The H1N1pdm "swine" influenza virus is closely related to the 2003 H5N1 "avian" influenza virus for which oseltamivir-resistance due to two individual point mutations, H274Y and N294S, is well known. However, the mechanism behind why these mutations actually inhibit successful binding of oseltamivir to neuraminidase is not well understood. Furthermore the mutations are actually located some distance from the drug-protein binding site, suggesting that the mutations may disrupt the actual drug binding process rather than just forming an inhospitable environment for drug-protein endpoint interactions.
In response, scientists from the University of Illinois and University of Utah teamed up to understand the mechanism behind mutation-induced drug resistance in influenza. The goal of this study was to model and simulate the binding of oseltamivir to H1N1pdm and H5N1 neuraminidase proteins, in order to a) understand at the molecular level what role these point mutations play in drug-resistance, and b) employ the findings to suggest strategies for designing drugs that can circumvent the virus' resistance mechanisms. An illustration of a representative simulated system is shown in Figure 1.
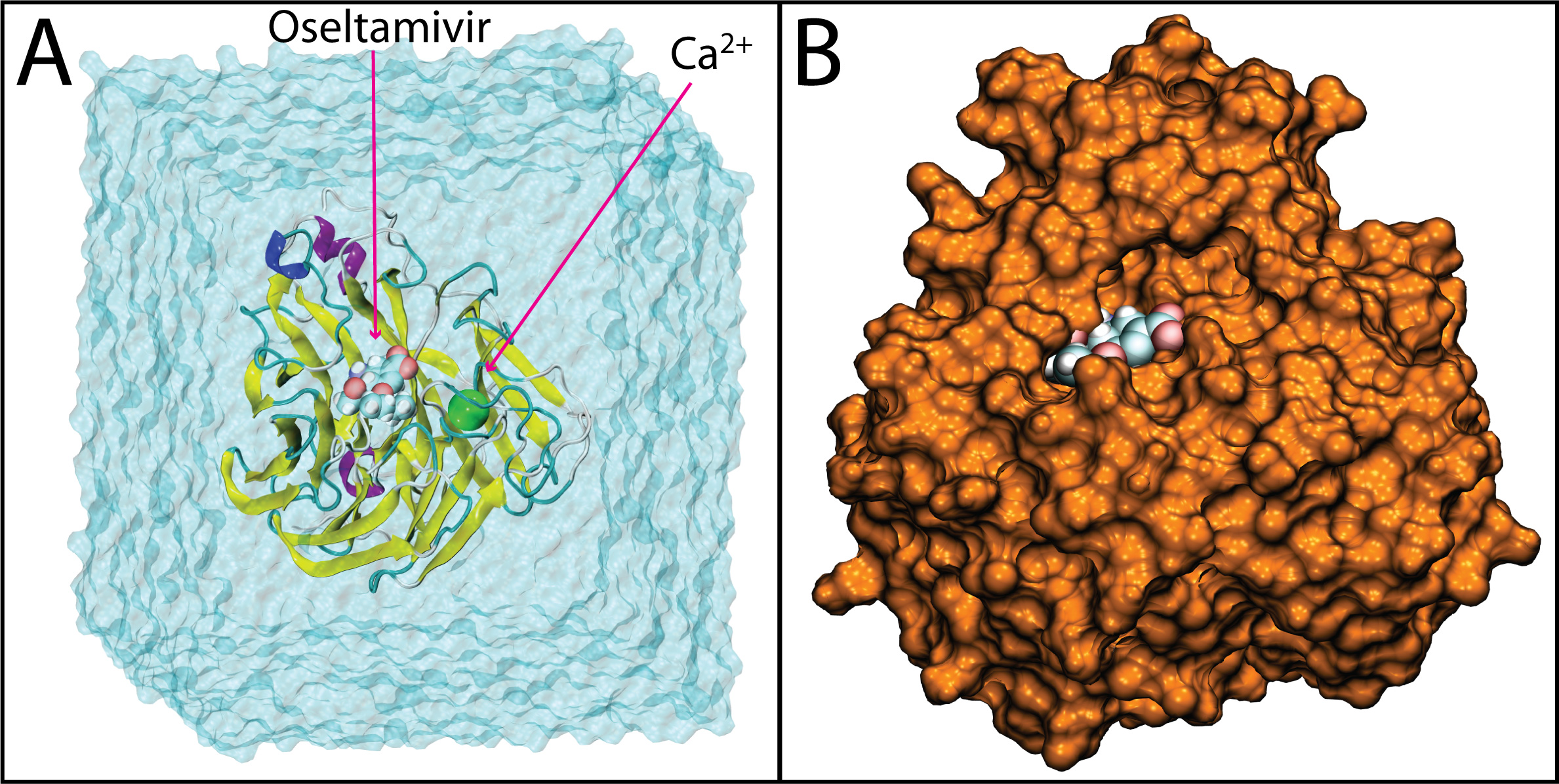
Figure 1. Drug bound systems simulated. Shown here is a representative example of H1N1pdm bound to oseltamivir. In A), the system is shown in the solvation box and with oseltamivir and the active site calcium ion. In B), oseltamivir is shown buried in the SA binding pocket of H1N1pdm, the latter rendered in surface view.
Molecular Modeling and Equilibration
The simulation systems involved neuraminidase proteins from the H1N1pdm and H5N1 influenza strains bound to oseltamivir. Both wildtype and two mutant variants (H274Y and N294S) were modeled for each neuraminidase, resulting in six systems being simulated. Equilibration simulations were first carried out for all six configuations, in order to relax each system and ensure stable drug protein binding. Analysis of drug protein interactions from these simulations revealed that a conserved network of hydrogen bonds (shown in Figure 2 for a representative system) tightly tether oseltamivir to its binding site on neuraminidase, even in the mutant systems. In the case of the H274Y mutant system, oseltamivir's pentyl group was observed to rotate slightly due to a loss of hydrophobic packing stability when compared to the wildtype system.
Figure 2. Network and occupancy of hydrogen bonds stabilizing oseltamivir in the SA binding pocket of wildtype and drug-resistant mutant avian H1N1pdm neuraminidases. A) Histograms of hydrogen-bond occupancies for interactions between oseltamivir and residues E119, D151, R152, R292, N347, and R371 across each simulation run. B) through D) Schematic views depicting the orientation of protein sidechains which form protein-drug hydrogen bonds. Hydrogen bonds in all three simulations were conserved for residues E119, D151, R292, and R371. The H274Y mutation was observed to disrupt hydrogen bonding to R152. Interestingly, residue 347, which distinguishes the binding pocket of H1N1pdm from H5N1, makes no contribution to the drug-protein hydrogen-bond network in the case of H1N1pdm.
Electrostatic Mapping
Examination of the equilibrium simulations alone, however, did not reveal any significant loss of binding stability in the mutant systems. It is well known that highly polarized drugs such as oseltamivir diffuse to their final binding sites through the so-called electrostatic steering phenomenon. That is, the surface charge of a protein likely exerts a significant influence on drug binding pathways. The surface electrostatic potential was therefore calculated over the entire equilibrium simulation trajectories. A GPU-accelerated version of the multilevel summation method was employed, and the use of this cutting edge technology reduced the time required to analyze a large trajectory from months to hours. The resulting electrostatic maps (see Figure 3 for a representative example), revealed a conspicuous negatively charged path on the surface of neuraminidase into its active site. Is this path a possible drug binding pathway?
Figure 3. Electrostatic surface potential of the sialic acid (SA) binding pocket of H1N1pdm and oseltamivir. Shown in A) and B) are closeup views of the SA binding pocket with drug bound H1N1pdm and avian H5N1 neuraminidase, respectively. The region of the binding pocket, where the drug binds, exhibits a negative potential (colored red), whereas the opening of the pocket is surrounded by a highly positive potential ring (colored blue). C) illustrates the electrostatic potential around oseltamivir. Shown are the "front" side facing the annulus of the binding pocket, and the "back" side facing the interior of the binding pocket. The simulations revealed the presence of a negatively charged funnel at the mouth of the binding pocket (colored in red) which may play a role in oseltamivir binding and unbinding. The electrostatic surface potentials were generated using the multilevel summation method (MSM) with color scale bars shown in insets.
Steered Drug Unbinding
Steered molecular dynamics (SMD) simulations were therefore performed to pull osteltamivir out of its binding site in order to probe possible unbinding and binding pathways. These simulations revealed that the key interaction stabilizing oseltamivir within its binding site were a series of hydrogen bonds between the drug's carboxyl group with Arg292 and Arg371 of neuraminidase. Application of force ruptures this critical interaction, causing the drug to rotate (see Figure 4) and thus freeing it to unbind. The simulations also revealed that the drug repeatedly unbound through a pathway which was tangential to the direction of pulling in the simulation. This pathway revealed in SMD followed the negatively charged pathway uncovered in the electrostatic calculations above. This observation suggested that there existed a preferential pathway through which oseltamivir could bind into neuraminidase guided by electrostatic steering. This pathway therefore, seems to function as an electrostatic binding funnel for the drug.
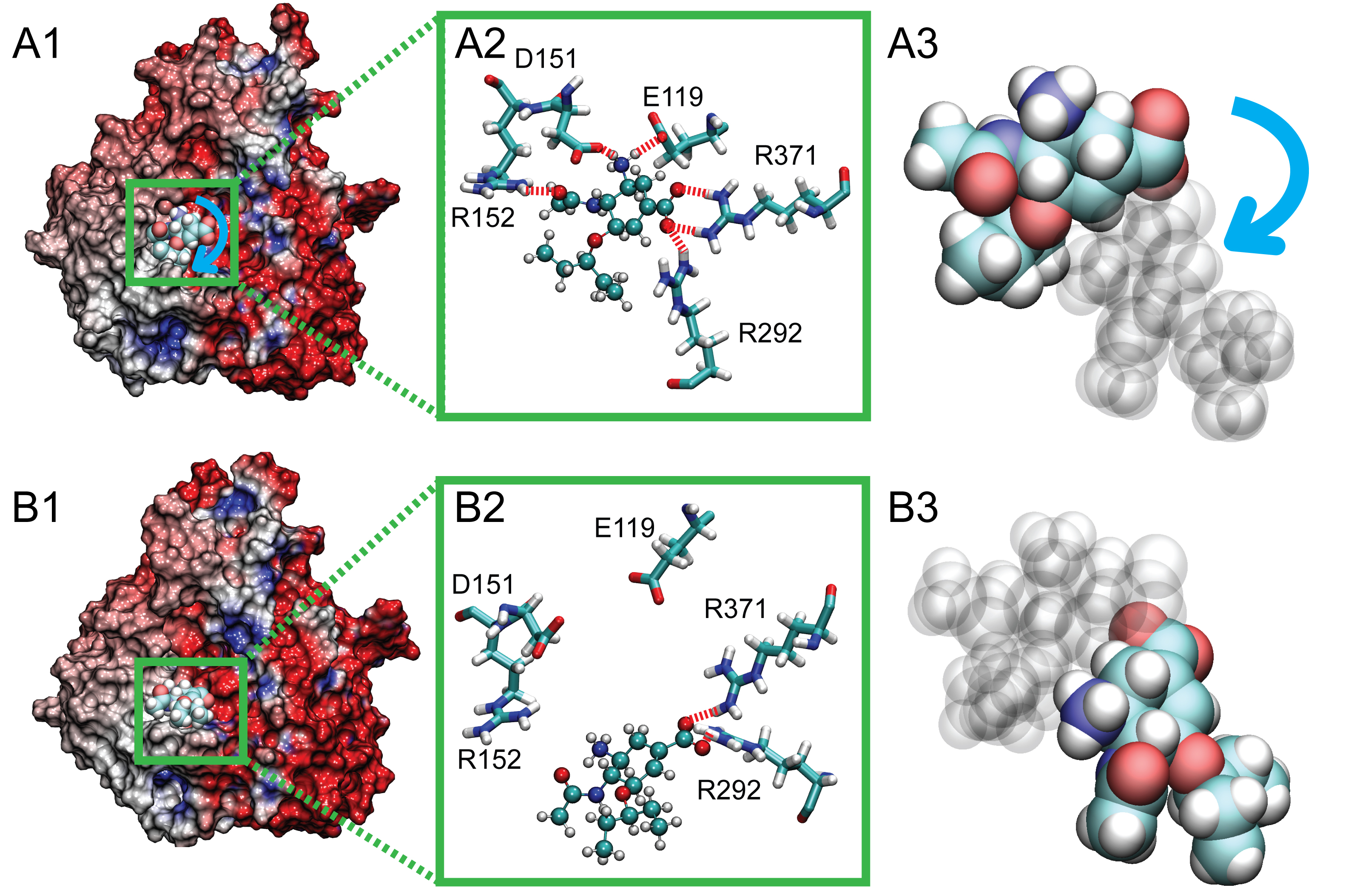
Figure 4. Rotated position of bound oseltamivir (arising at 7.5ns) in comparison with its stable equilibrium position, shown with the electrostatic surface potential of the protein. In A1, the stable binding pose of oseltamivir is shown prior to application of force in simSMD1, with stable hydrogen bonds to E119, D151, R152, R292, and R371 (shown in A2). After 7.5 ns, however, oseltamivir adopted a new binding pose (shown in B1), where hydrogen bonds with E119, D151, and R152 are ruptured, leaving only stable interactions with R292 and R371 (shown in B2). The blue arrow in A3 denotes the orientation change during the drug's rotation in the binding pocket. The rotated bound oseltamivir shown in B1, B2 and B3 served as the starting point for simulations simFEQ1-10.
Elucidating a Drug Binding Funnel
While SMD simulations can suggest unbinding pathways, one must make an assumption that the reverse process is also viable. In order to test this hypothesis, the researchers performed additional equilibrium simulations which were able to capture both the unbinding and rebinding of oseltamivir through the electrostatic funnel in the absence of force (see Figure 5). These simulations therefore, offered concrete evidence that the binding funnel probed in the SMD simulations above were not a result of chance, but had actually elucidated a novel binding pathway. The two known drug resistant mutations are located proximal to this binding pathway, and it is therefore possible that the mechanism of drug resistance arises from their disruption of oseltamivir passage through this funnel.
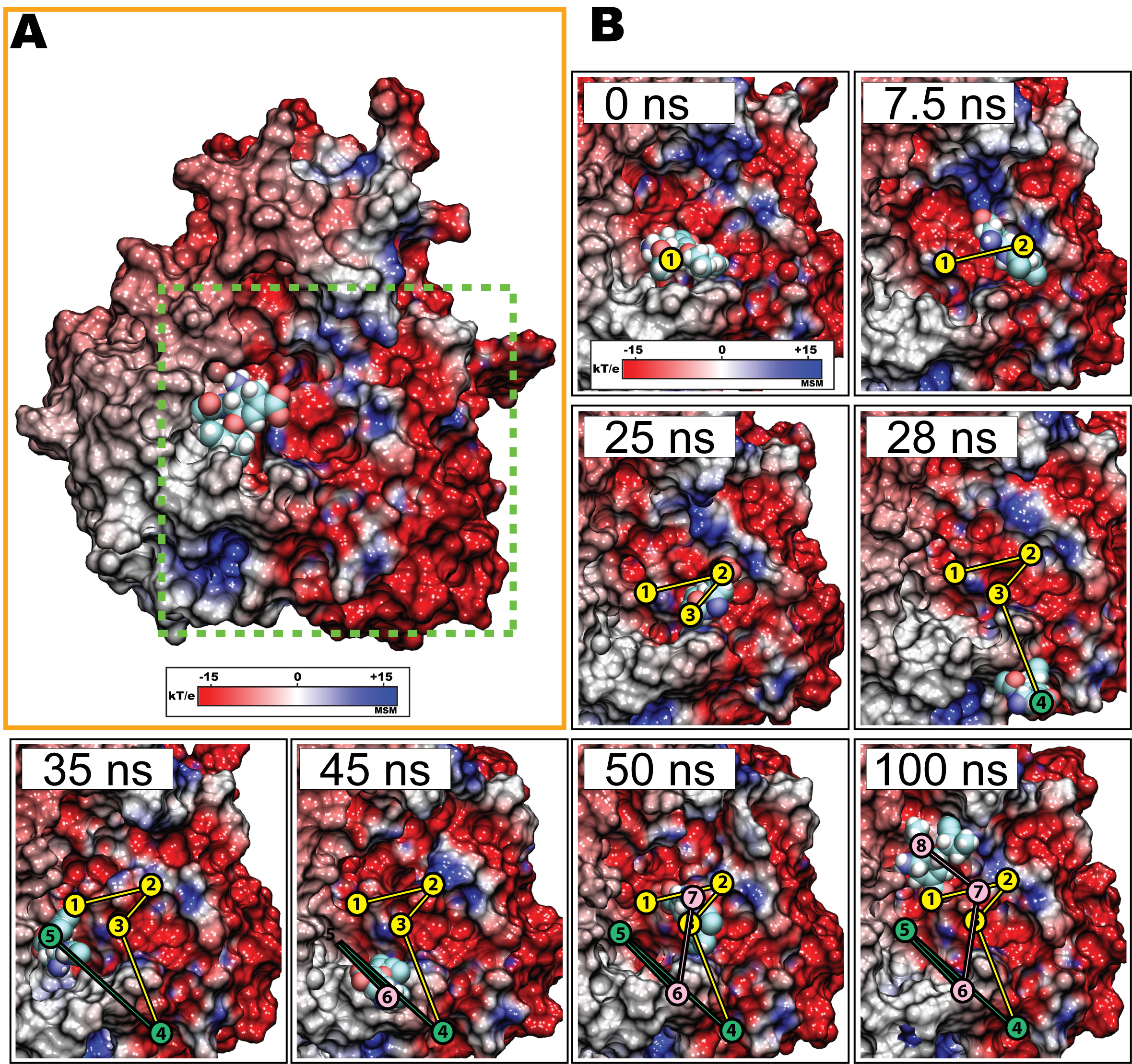
Figure 5. Escape and rebinding of oseltamivir through the electrostatic binding funnel in H5N1 neuraminidase during simulation. Shown here are snapshots in which oseltamivir first diffuses out of the SA binding pocket through interaction with the electrostatic binding funnel (see Figure 3) within the first 25 ns of simulation. Between 28 and 35 ns, oseltamivir, now free from neuraminidase, approaches the periphery of the binding pocket away from the binding funnel, but is prohibited from entering due to electrostatic repulsion (45 ns). However, between 45 and 50 ns, oseltamivir reapproaches and rebinds with neuraminidase through the same, i.e., the electrostatic, binding funnel, adopting a stable position within the SA binding pocket through hydrogen bonds with Y406, R292, D151, E119, R118.
Conclusion
Our results demonstrate not only a viable pathway for the binding of an antiviral drug, but also how the disruption of such a pathway from known point mutations can function as the basis for resistance to the front line anti-influenza drug oseltamivir. The discoveries from this study should pave the way for designing drugs which can bind stably to flu neuraminidase despite disruption of H274Y hydrophobic and N294S pathway interactions.
Post Publication Review
This article was suggested as a Must Read in Faculty 1000 with the following evaluation:
This study highlights, using molecular dynamics (MD) simulations, a new method of understanding the binding of the antiviral drug oseltamivir (Tamiflu) to neuraminidase. In contrast to equilibrium studies that examine the bound (or unbound) states, the authors investigate the pathway of drug binding and its relationship to resistance. They reveal a novel finding that the kinetics of the drug appear to be controlled by electrostatic funnels in the enzyme. This may help discover new allosteric mechanisms of how resistance arises due to mutations that are distant from the active site and how these modulate drug binding. The authors perform MD simulations totalling ~600ns to study the interactions between Tamiflu and neuraminidase in its wild-type and mutant states. These combine equilibrium and pulling simulations. The authors suggest that electrostatics along the binding cavity appear to play a critical role in enabling the ligand to enter and bind, and they show how local conformational modulation is critical to this process. Indeed, a combination of equilibrium and pulling experiments shed light on the various mechanisms that no doubt underpin the effects of resistant mutations. This is the first such finding and while it will be interesting to see how the other mutations map onto this model, nevertheless it opens the way for designing new drugs that will exploit electrostatic funneling. The study adds to our growing understanding of the subtle and complex landscapes that modulate intermolecular interactions and points to the need for examining, in detail, the actual process of ligand entry/exit/egress into/from the binding site [1,2]. In addition, such findings are critical for the development of methods to understand and predict how resistance may likely arise in response to the nature of the drug as well as to the protein architecture [3].
References:
[1] Colizzi et al. J Am Chem Soc 2010, 132:7361-71 [PMID:20462212].[2] Baron et al. J Am Chem Soc 2010, 132:12091-7 [PMID:20695475].[3] Frey et al. Proc Natl Acad Sci U S A 2010, 107:13707-12 [PMID:20643959].
Movies
Unbinding and rebinding of oseltamivir (Tamiflu) into the active site of neuraminidase through the charged binding funnel (mpeg-4, avi).
Related publications
-
Publications Database Molecular dynamics simulations suggest that electrostatic funnel directs binding of Tamiflu to influenza N1 neuraminidases. Ly Le, Eric H. Lee, David J. Hardy, Thanh N. Truong, and Klaus Schulten. PLoS Computational Biology, 6:e1000939, 2010. (13 pages). -
Publications Database Molecular modeling of swine influenza A/H1N1, Spanish H1N1, and avian H5N1 flu N1 neuraminidases bound to Tamiflu and Relenza. Ly Le, Eric H. Lee, Klaus Schulten, and Thahn Truong. PLoS Currents: Influenza, 2009 Aug 27:RRN1015, 2010. (9 pages).
Investigators
- Eric H. Lee
- Ly Le
- David Hardy
Collaborators
- Thanh N. Truong (University of Utah)
Page created and maintained by Eric H. Lee and David Hardy.


