Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
image size:
91.7KB
made using VMD
image size:
215.8KB
made with VMD
image size: 468.6KB
image size:
397.0KB
made using VMD
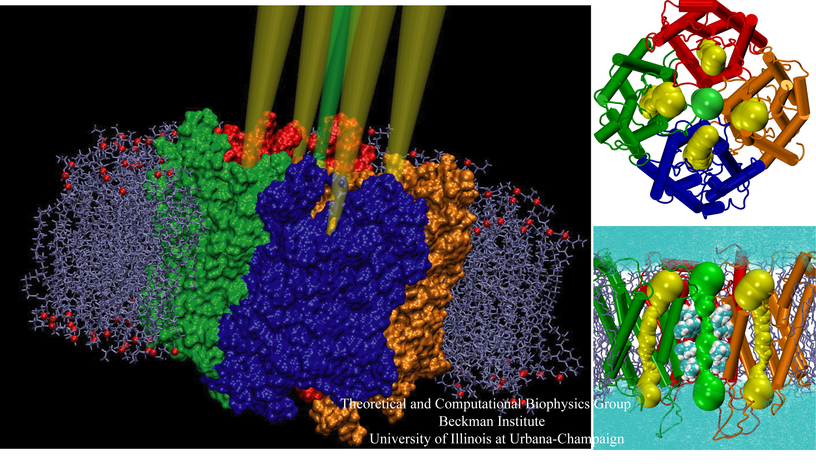
image size:
501.0KB
made using VMD

image size:
285.6KB
made using VMD
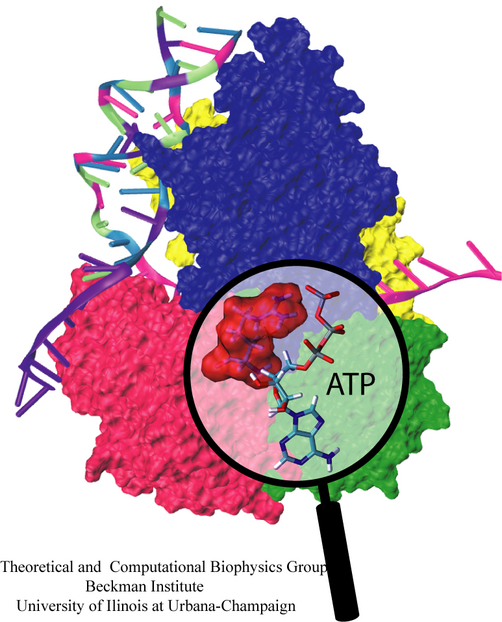
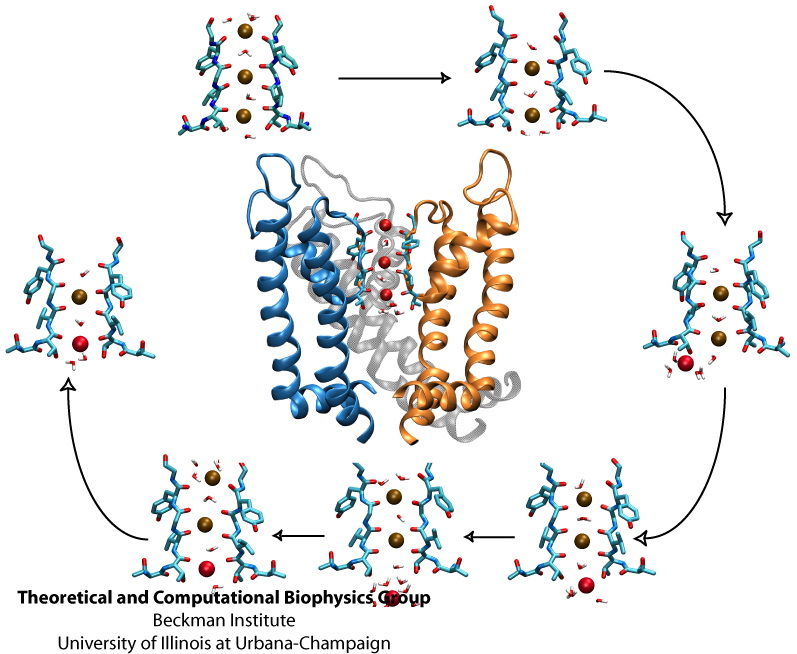
image size:
249.5KB
made with VMD
image size:
300.4KB
made with VMD


image size:
112.7KB
image size:
194.1KB
made with VMD

image size: 87.6KB

image size:
49.3KB
made with VMD
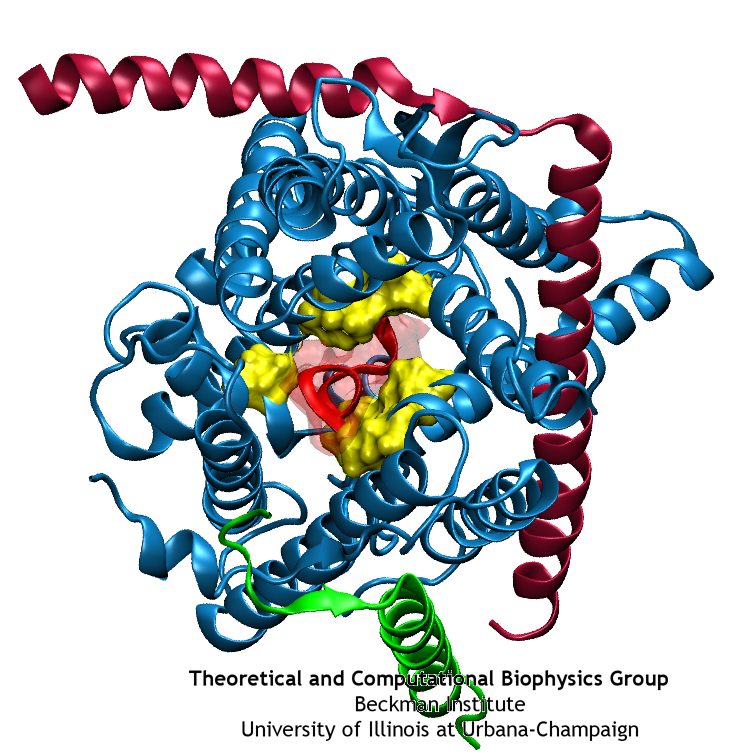
image size:
40.4KB
made with VMD
Anyone who has attempted to fit a long piece of thread through a needle's eye realizes how difficult fitting something so small and flexible into such a small hole can be. Yet this action is carried out every second in every living cell. Flexible polypeptides, proteins, often have to cross a cellular membrane to get to their correct location, whether that location is an organelle within the cell or even outside of it. To accomplish this, they are pushed through a protein pore in an unfolded conformation much like a long string. The channel that accepts the string-like proteins, the protein translocon, allows only certain proteins to pass, while restricting access to molecules even much smaller than the macromolecular proteins. As reported in a recent publication, computer simulations using the molecular dynamics program NAMD helped to answer the question of how such a small channel could achieve this feat, demonstrating how the channel itself can be flexible yet resilient during a protein-crossing event and also elucidating in part how it can maintain such tight control over what is permitted to cross. For more information, see our Protein Translocation website.
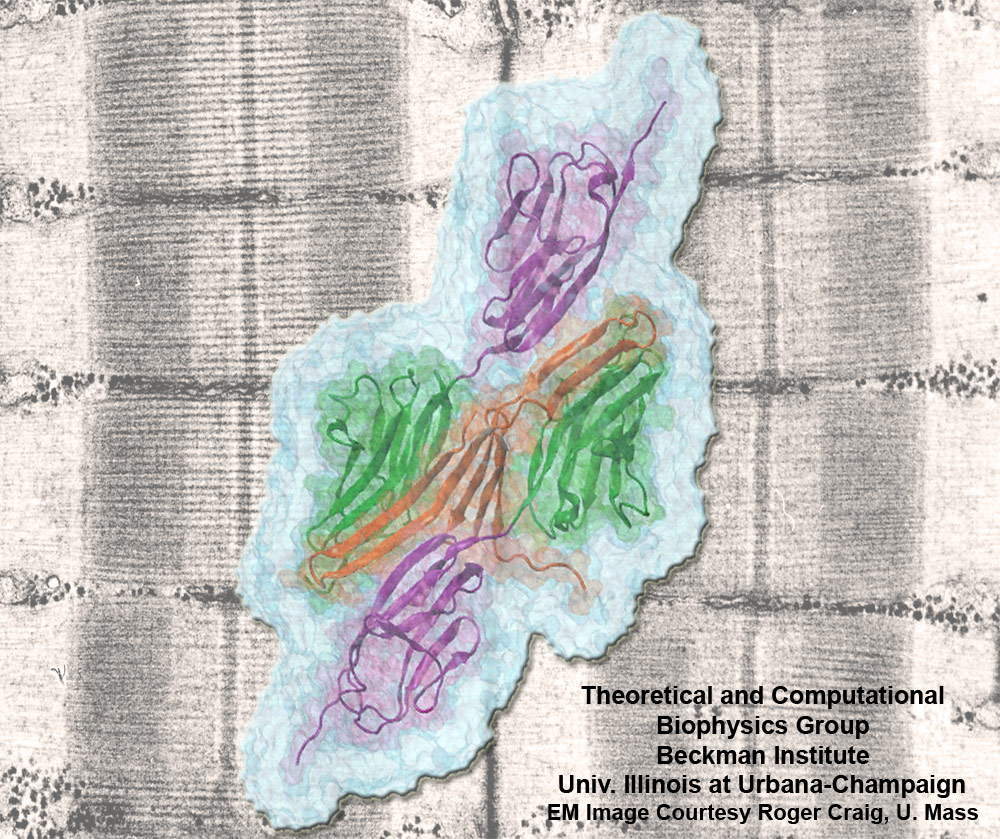
image size:
301.5KB
made with VMD
Muscle fibers, through their so-called thick and thin filaments, contract and extend in doing their work. To render the fibers elastic and protect them from overstretching, the thick filaments are connected through a long and thin elastic protein, titin, to the base of the fibers. Titin, by far the longest protein in human cells, is a molecular bungee cord and, like such cord, must be affixed firmly to the base. How this is done was a mystery until crystallographers took the first atomic resolution image of the system: it turns out that two titins are spliced together at their ends like ropes. The splicing involves a third small protein, the titin-telethonin-titin system forming a U. The U apparently is thrown over a bollard-like cellular structure to hold the thick filaments much like boats are held by bollards and ropes at their mooring place. The crystallographers teamed up with computational biologists to investigate the mechanical strength of the titin - telethonin - titin cord by means of molecular dynamics simulations using NAMD. As reported recently, the cord has great mechanical strength due to an extended network of hydrogen bonds between beta-strands, common structural features in proteins, that in the present case form a sheet extending through all three proteins. This discovery explains how living cells can splice cellular proteins together through a system of hydrogen-bonded beta-strands that extend through several proteins. Interestingly, such beta-strands were seen previously in cases of diseases like Alzheimers where the feature leads, however, to pathological assembly of proteins. What needs to be understood now is how the telethonin glue is applied only to the right spots in the cell and how the cells prevent telethonin from splicing together the wrong proteins. For more information visit our titin-telethonin web page.
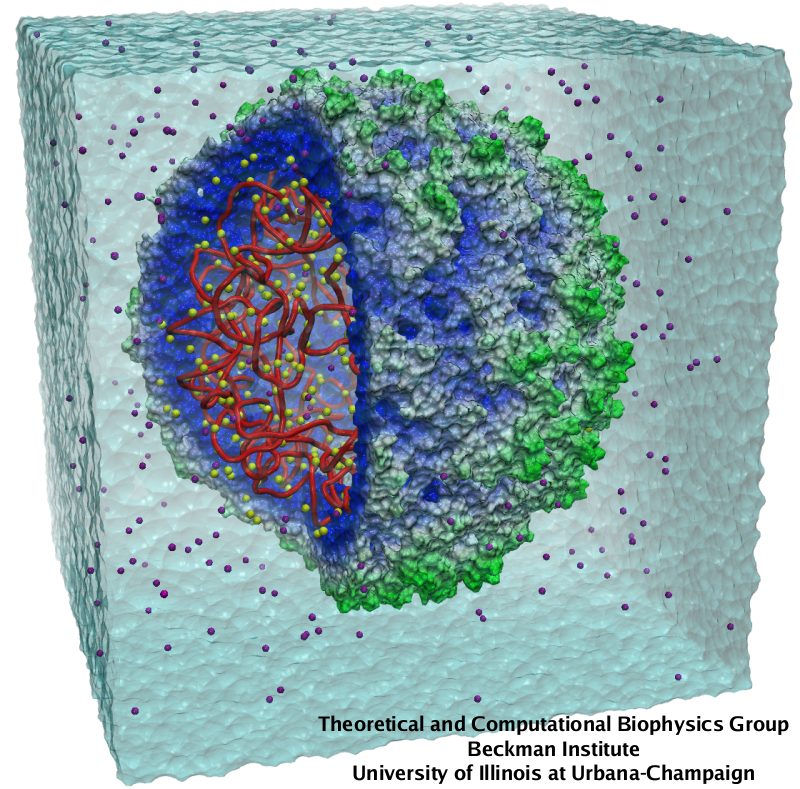
image size:
150.4KB
made with VMD
Viruses, the cause of many diseases, are the smallest natural organisms known. They are extremely primitive and parasitic such that biologists refer to them as "particles", rather than organisms. Viruses contain in a protein shell, the capsid, their own building plan, the genome, in the form of DNA or RNA. Viruses hijack a biological cell and make it produce from one virus many new ones. Viruses have evolved elaborate mechanisms to infect host cells, to to produce and assemble their own components, and to leave the host cell when it bursts from viral overcrowding. Because of their simplicity and small size, computational biologists selected a virus for their first attempt to reverse-engineer in a computer program, NAMD, an entire life form, choosing one of the tiniest viruses for this purpose, the satellite tobacco mosaic virus. As described in a recent report, the researchers simulated the virus in a small drop of salt water, altogether involving over a million atoms. This provided an unprecedented view into the dynamics of the virus for a very brief time, revealing nevertheless the key physical properties of the viral particle as well as providing crucial information on its assembly. It may take still a long time to simulate a dog wagging its tail in the computer, but a big first step has been taken to simulate living organisms. Naturally, this step will assist modern medicine (more on our satellite tobacco mosaic virus web page).
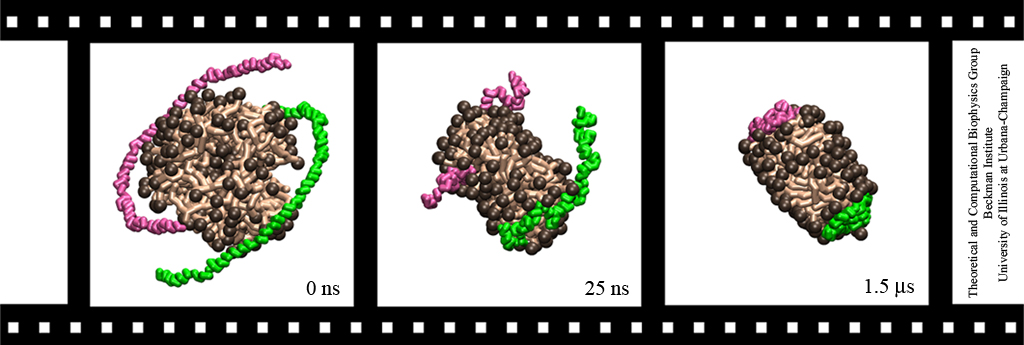
image size:
153.6KB
made with VMD
Lipoproteins are protein-lipid particles which circulate in the blood collecting cholesterol, fatty acids, and lipids. Low levels of one such lipoprotein particle, called high-density lipoprotein (HDL) or "good cholesterol", has been implicated in the increased risk of coronary heart disease. The ability of lipoproteins to transport lipid and cholesterol through the blood is amazing since these types of particles are not generally soluble in blood plasma. However, when HDLs assemble, proteins wrap themselves around the lipids and cholesterol, shielding the lipid tails from the aqueous environment. Native HDL exhibit a variety of shapes and sizes, for example forming a discoidal particle. Conventional high-resolution imaging techniques, such as NMR and X-ray crystallography, cannot resolve how lipid and cholesterol are being accommodated by HDL, but the assembly and geometry of HDL discs can be captured using computer simulations. Unfortunately, the long time scales required for HDL assembly was a major stumbling block. Now a new simulation method, coarse-grained modeling in conjunction with the molecular dynamics program NAMD, has permitted the simulation of HDL assembly as recently reported. The simulations show that lipids quickly aggregate into a bilayer from their initial spherical "micelle" shape and that the two proteins subsequently attach to either side forming a belt surrounding the lipid core. For more information see HDL & nanodisc and coarse-grained modeling.
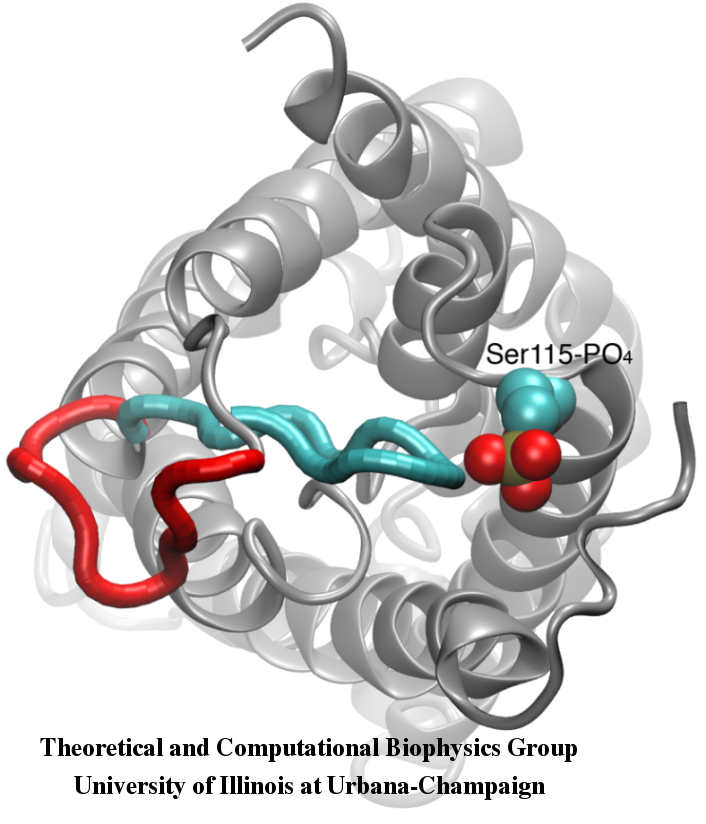
image size:
258.5KB
made with VMD
Your favorite flower pot would not survive a weekend in your office without watering, if it wasn't for a sophisticated cellular mechanism evolved in land plants to conserve water under drought conditions. Water exchange between cells and their environment is facilitated by a group of highly specialized membrane proteins called aquaporins. Although present in all life forms, plants are particularly dependent on their function. While in most species these channels function as always-open "cellular pipes" allowing water in and out of the cell, in plants they evolved into "cellular faucets" whose water permeability can be controlled by the cell. Nearly all plant aquaporins can be gated in response to drought or even flooding conditions, through basic biochemical signals, e.g., phosphorylation and change of pH. A recent Nature paper reporting a collaborative study between crystallographers who succeeded in solving the first structure of a plant aquaporin from spinach, and modelers provides the most detailed view of the mechanism of gating for a membrane channel. Molecular dynamics simulations of the channel performed by NAMD reveals a dual gating mechanism in which phosphorylation of certain protein residues unleashes a long cytoplasmic loop that physically blocks water access to the pore. More information on aquaporin research can be found here.
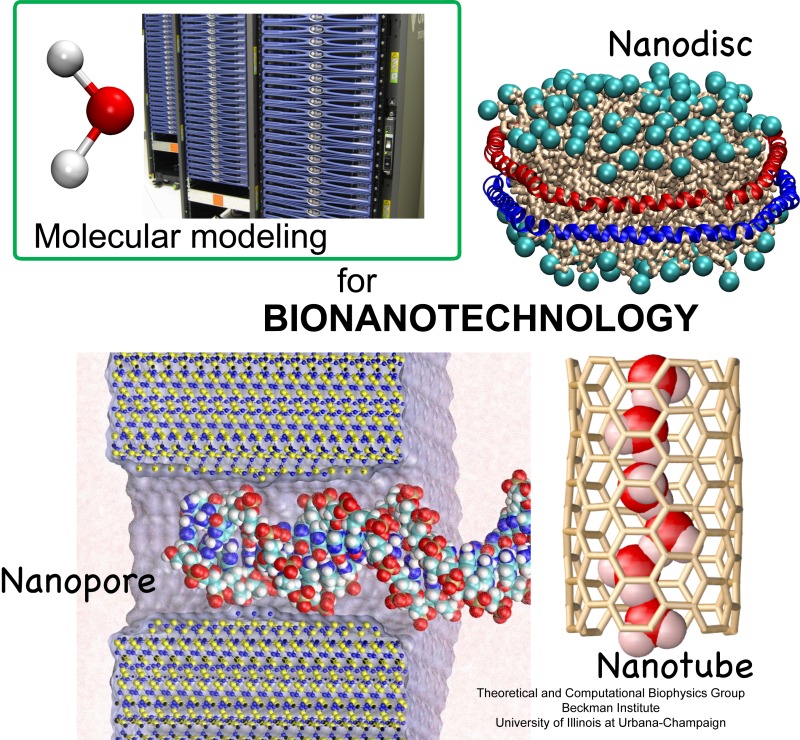
image size:
225.3KB
made with VMD
Molecular modeling with NAMD (NAnoscale Molecular Dynamics) promises to become a key methodology for research and development in bionanotechnology. Molecular modeling provides nanoscale images at atomic and even electronic resolution, predicts the nanoscale interaction of yet unfamiliar combinations of biological and inorganic materials, and can evaluate strategies for redesigning biopolymers for nanotechnological uses. The methodology's value has been reviewed for three uses in bionanotechnology. The first involves the use of single-walled carbon nanotubes as biomedical sensors where a computationally efficient, yet accurate description of the influence of biomolecules on nanotube electronic properties and a description of nanotube - biomolecule interactions were developed; this development furnishes the ability to test nanotube electronic properties in realistic biological environments (see Dec 2005 highlight). The second case study involves the use of nanopores manufactured into electronic nanodevices based on silicon compounds for single molecule electrical recording, in particular, for DNA sequencing. Here, modeling combining classical molecular dynamics, material science, and device physics, describes the interaction of biopolymers, e.g., DNA, with silicon nitrate and silicon oxide pores, furnishes accurate dynamic images of pore translocation processes, and predicts signals (see Nov 2005 and Oct 2004 highlights). The third case involves the development of nanoscale lipid bilayers for the study of embedded membrane proteins and cholesterol. Molecular modeling tested scaffold proteins, redesigned lipoproteins found in mammalian plasma that hold the discoidal membranes in shape, and predicted the assembly as well as final structure of the nanodiscs (see Feb 2005 highlight). In entirely new technological areas like bionanotechnology qualitative concepts, pictures, and suggestions are sorely needed; the three exemplary applications document that molecular modeling can serve as a critical "imaging" method for bionanotechnology, even though it may still fall short on quantitative precision.
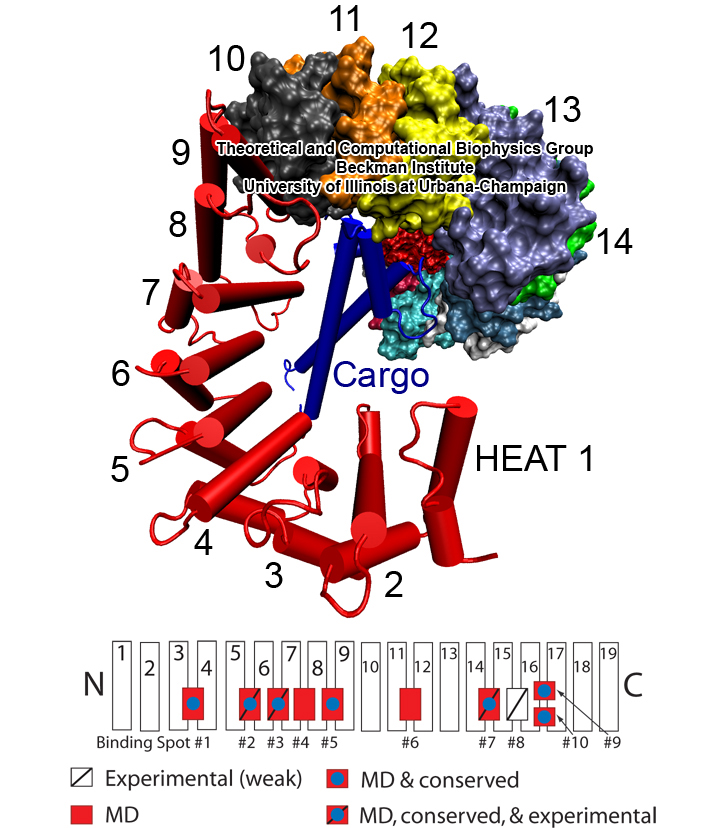
image size:
360.8KB
made with VMD
Eukaryotic cells envelop their genetic material in the cell nucleus whose boundary contains numerous pores. Only small molecules can pass through these nuclear pores unhindered. For all larger ones, passage is highly selective and controlled. The control involves import and export proteins (transport receptors) that load and release cargo on the proper side of the nucleus upon interaction with signaling proteins. Researchers are presently solving the structure of the nuclear pore and its transport receptors with increasing resolution, and the first atomic level investigation into the mechanism of nuclear pore selectivity has recently been reported [paper]. The study inspected the interaction between the transport receptor importin-β with key nuclear pore proteins that appear disordered near the center of the pore and contain characteristic phenylalanine-glycine sequence repeats. Molecular dynamics simulations using NAMD and analyzed using VMD revealed a key insight into the selectivity mechanism. The simulations showed that the key sequences of the repeat proteins interact strongly with certain spots on the surface of importin-β. The study confirmed spots that had previously been identified experimentally and, moreover, found numerous binding spots not yet seen in experiment. Further experiments and simulations promise an understanding of the selectivity of entry and exit from the nucleus, a key element of the cell's genetic control. For more information see our nuclear pore complex webpage.


