Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
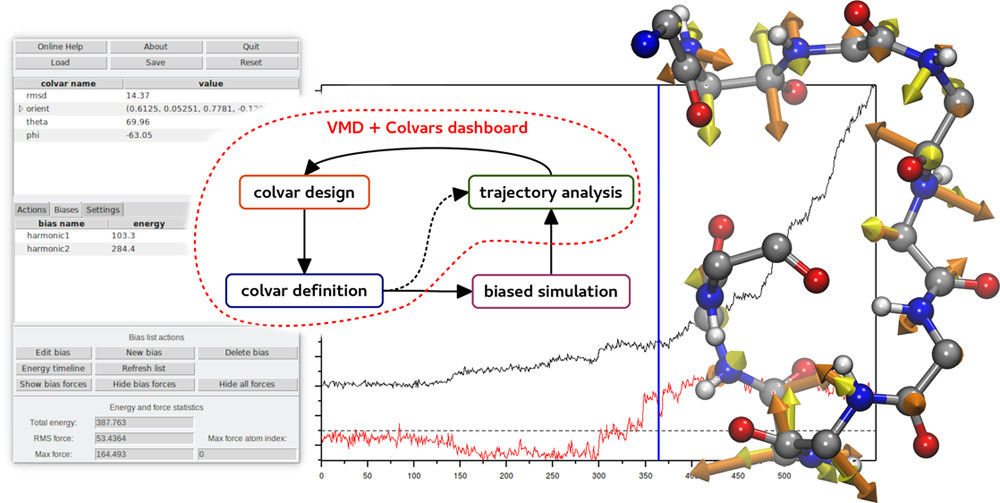
Collective Variables (COLVARS) are an indispensable component of modern molecular simulations allowing one to execute enhanced sampling
and free energy calculations along complex reaction coordinates. The Collective Variables Dashboard allows for real-time, seamless
exploration of molecular structures and trajectories. It arises from the integration of the
COLVARS Module with VMD, augmented with a graphical interface
offering interactive workflows for the design and analysis of collective variables. The Dashboard allows a priori design of collective
variables for simulations as well as analysis of any type of simulation trajectories. In addition, it provides an efficient way to intuitively
explore the space of likely collective variables, validate them on existing data, and use the resulting collective variable definitions
directly for additional simulations. Finally, it visualizes the energies and forces to help analyze or plan simulations.
Learn more about the Dashboard in a recent article, through
a 5-minute video, or this lecture.

The COVID19 pandemic has resulted in massive pain and distress to all aspects of human life. A key viral element for effective infection is the surface protein, spike (the Corona), which has been a major target for drug development against the virus. Using multimicrosecond simulations with NAMD and analyzed with VMD, we show that glycosylations of the spike, which are known to shield the protein from most antibodies, also equip the protein with a larger range of motion, thus making it more effective in search of human receptors. Read more about the story in this U. of I. News and in Proceedings of the National Academy of Sciences.
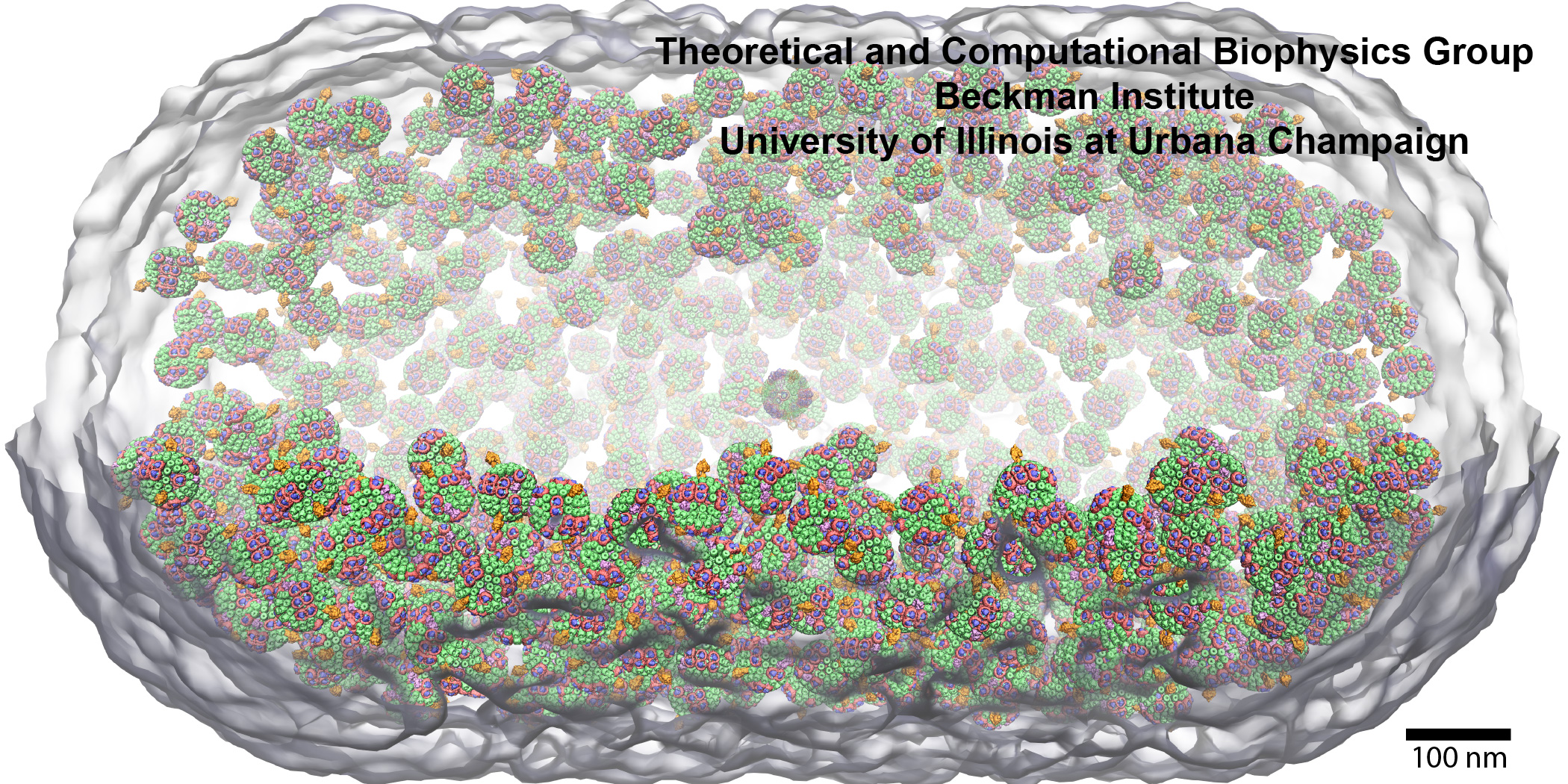
Paraphrasing Douglas Adams, cells are "big ... vastly, hugely, mind-bogglingly big"
—
especially when looked at it in atomistic detail. Telling the story of a cellular process often requires a multitude of scales, spanning electronic, atomic, protein, and organelle sized structures before integrating them into an entire cell. The story of how light powers life is one such story
—
in fact, the oldest story of humanity, now retold in a multi-scale atoms-to-cell computational model built upon a decade-long experimental collaboration.
Visualization of such cell-scale models is recently made possible by the addition of instancing support for molecules into VMD.
The new VMD instancing feature permits molecular scientists to create an arbitrary number of graphical copies or instances of user-selected molecular components, proteins, or entire complexes, thereby facilitating cell-scale visualization. The set of instances to be visualized are described by assigning a set of transformation matrices and graphical representations that VMD uses to the complete visualization.
VMD's instancing capabilities are illustrated in the cover design of
Molecular Mechanisms of Photosynthesis
(Wiley, 2021)
by
Robert Blankenship.
The structural model
employed for the cover is that of a low light adapted
Rba. sphaeroides cell featuring 985 chromatophores
with a radial distribution consistent with cryoEM tomography.
Combined with
award winning advances in multi-scale simulation techniques,
these visualization methods enable the quantification of cellular processes
from individual atoms to organizing principles.
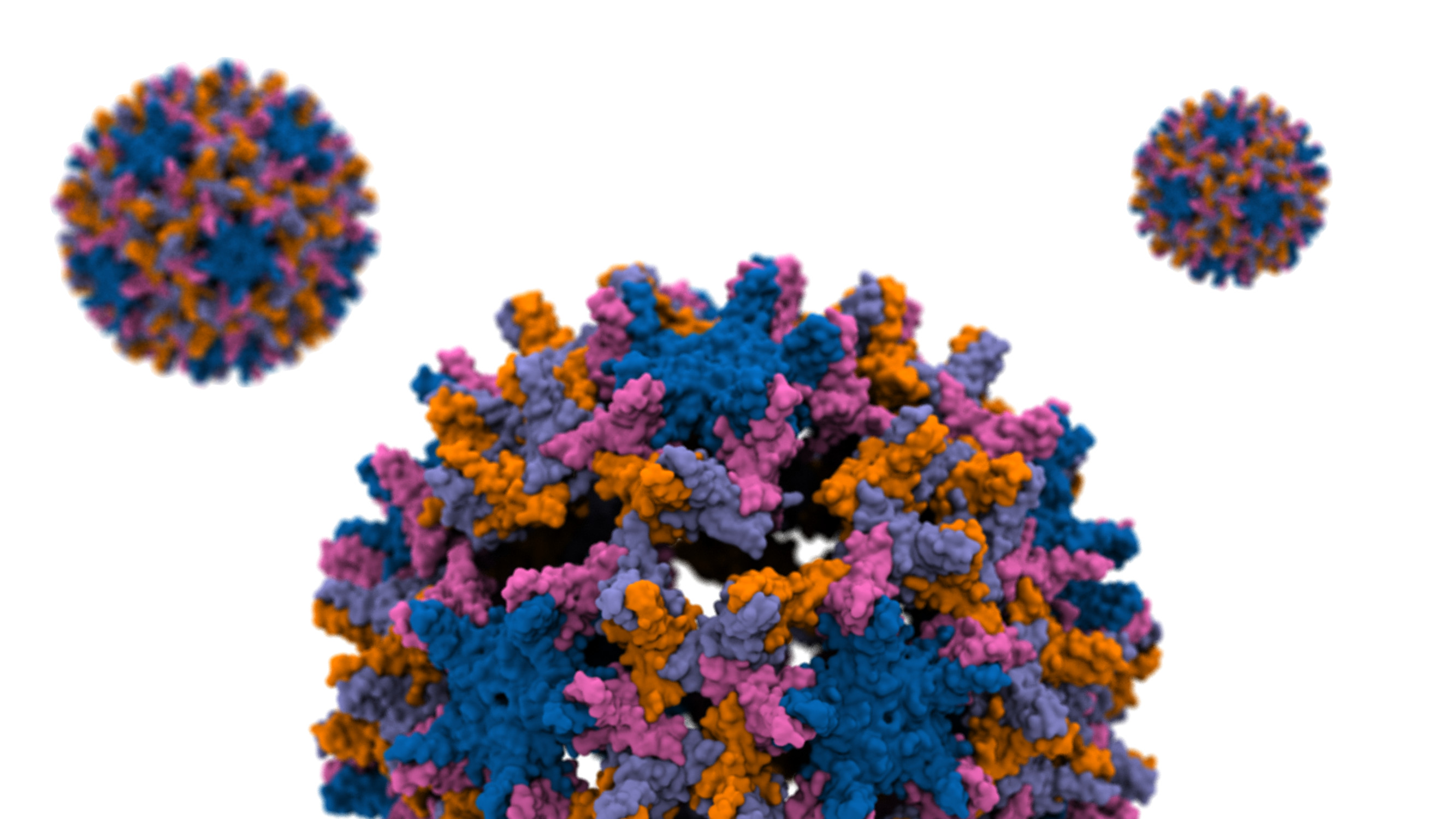
The hepatitis B virus (HBV) is one of the smallest human pathogens that infects liver cells chronically affecting ~250 million people globally. The breaking of the HBV capsid leads to the release of its genetic material into the host cell and therefore a key step in its infection. The capsid consists of hundreds of identical protein blocks. Using molecular dynamics simulations with NAMD we devised a mechanical stress to study the process of capsid disassembly in a simulation composed of millions of particles. Analyzing the resulting capsid cracks by VMD we show that the cracks mainly occur within a particular (hexameric) protein arrangements. More importantly, only a small subset of the capsid proteins are engaged in initiating the disassembly. The majority of such hot-spot residues are conserved through evolution, hinting at their importance in the viral infection process, probably by destabilizing the capsid to the right degree. Read more in the Beckman Institute press release and the published article in Proceedings of the National Academy of Sciences.
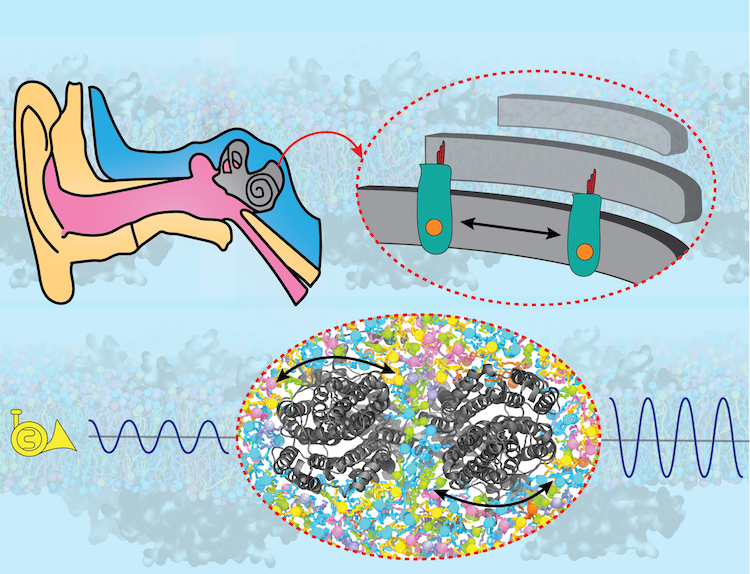
Sound amplification is a key step in hearing, without which the brain would not detect the majority of sounds generated in our surroundings. The key protein responsible for sound amplification is a motor protein called prestin in the inner ear. After decades of research, high-resolution structures of prestin were recently obtained by cryo-EM, revealing how the protein's shape change in response to electrical signals "shakes" the hair cells in the inner ear, thereby amplifying the sound to the level that is detectable by neurons. In collaboration with structural biologists at Oregon Health and Science University who solved the protein structures, and employing NAMD and VMD, the Center's researchers shed light on how prestin transmits its structural change to the cellular membrane and thereby shakes the hair cell in our auditory system. Read more about the study in Cell and in the U. Illinois Biophysics Press Release.


