Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001

image size:
86.3KB
made with VMD
Motor proteins are fascinating cellular machines that convert chemical energy into mechanical work. They are employed in a wide range of cellular functions like muscular contraction, transportation of proteins and vesicles, and cell motility. Myosin VI is an example of a motor protein. It "walks" along actin filaments (kind of like cellular highways), performing tasks such as delivering materials across the cell. Primarily, myosin VI functions as a dimer (i.e., two myosin VI proteins are associated and form a functional complex), but the structure of the myosin VI dimer, particularly how a myosin VI associates with another one, is still debated. Teaming up with experimentalists, computational biologists investigated how two myosin VI assemble and pull their cargo together. The investigation, reported recently, focused on a segment of myosin VI that forms a long, rigid alpha-helix that is notably decorated with a distinct rings of positively and negatively charged amino acids. Carrying out single-molecule experiments along with molecular dynamics simulations using NAMD, it was found out that two myosin VI proteins attract each other electrostatically through the charge-ring proteins, shifting them such that the oppositely charged amino acids from different helices face each other. More information can be found on our motor protein website.
2.7 release
of NAMD
includes GPU acceleration,
downloadable binaries
for Linux clusters with InfiniBand networks,
grid-based forces and ..." />

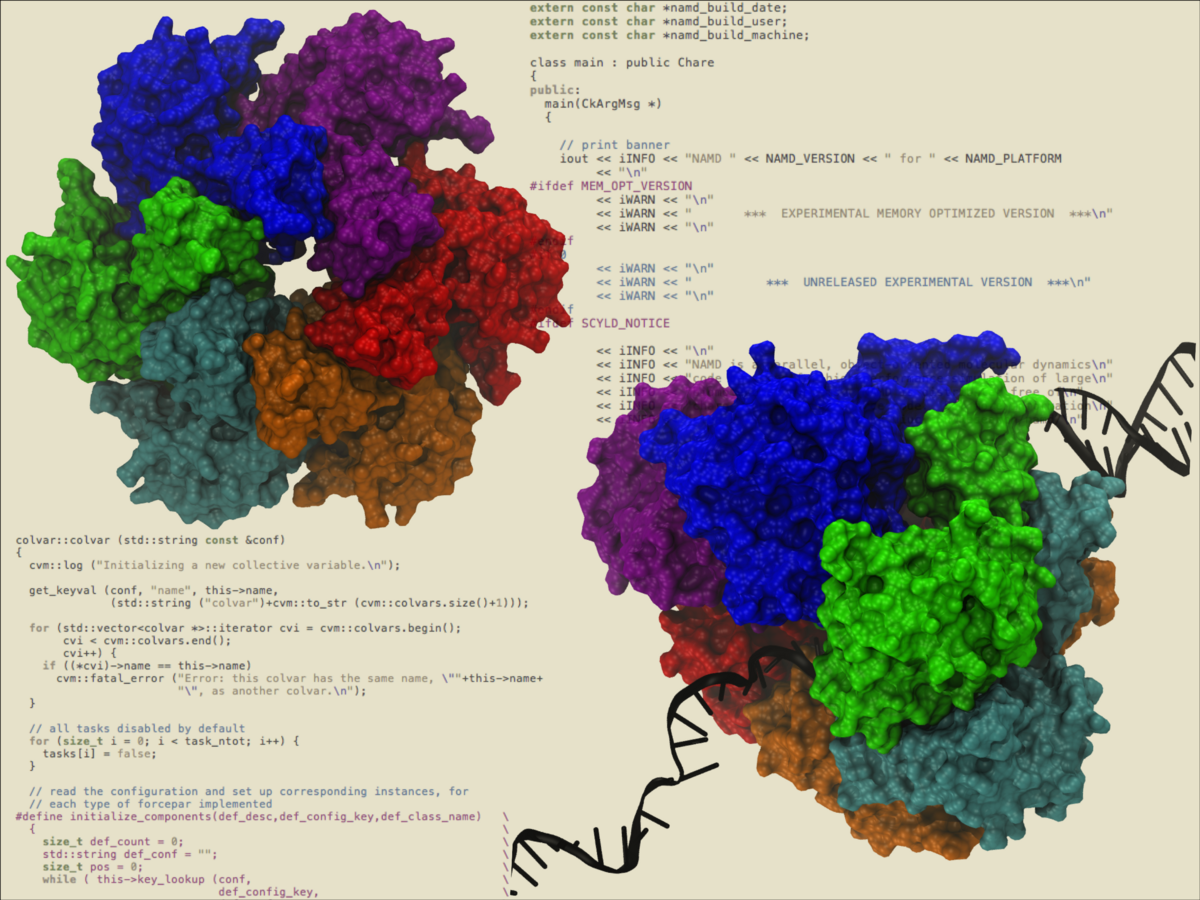
image size:
994.9KB
made with VMD
The eagerly awaited 2.7 release
of NAMD
includes GPU acceleration,
downloadable binaries
for Linux clusters with InfiniBand networks,
grid-based forces and extra bond/angle/torsion conformational restraints
for molecular dynamics flexible fitting (MDFF), and a major
enhancement of both alchemical and conformational free energy methods.
Alchemical calculations gradually alter the chemical structure of a
molecule during a simulation, monitoring the consequences of creating and
deleting atoms via either free energy perturbation or thermodynamic
integration.
A soft-core correction may be employed to
circumvent singularities when atoms are created.
Conformational calculations can probe structural rearrangements along
an arbitrary number of collective variables,
including distances between atoms or groups of atoms, distances projected
along an axis or in a plane, angles, torsions, eigenvectors,
gyration radii, coordination, root mean-square displacements, orientations,
and alpha-helicity. New variables can be introduced without recompilation.
Free energy surfaces or potentials of mean force can be constructed using
metadynamics, the adaptive biasing force method, umbrella sampling, and
steered molecular dynamics.
The collective variables capability of NAMD
has allowed the simulation of the
illustrated hexameric helicase. In the simulation, a single strand of DNA
passes through the central core while the hexamer translocates or "walks"
along the DNA, a mechanical process driven by the energy of ATP hydrolysis.
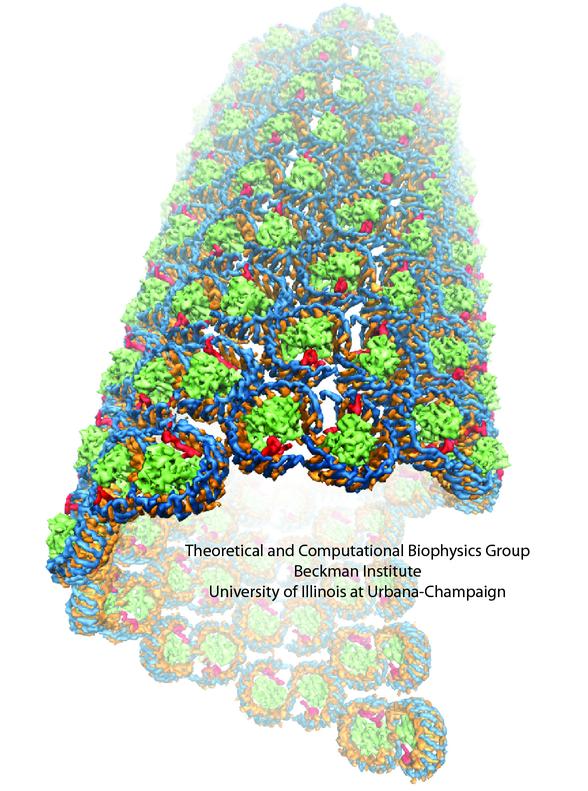
The process of photosynthesis fuels life on Earth. Its first step is capturing the energy in sunlight. Light-capturing proteins in photosynthetic organisms are often seen closely crowded together in the cellular membrane, forming hundred nanometer-sized patches. Such "photosynthetic membranes" can be flat or spherical, depending on bacterial species (see the October 2007 highlight on Life's Solar Battery, the August 2010 highlight on Bacterial Solar Energy Engineering, and a recent review); in case of a certain mutant bacterium the membrane forms the cylindrical surface of a rod. This membrane is actually an ideal case for scientific investigation, since it contains only one type of protein complex organized in an orderly fashion, such that placement of all proteins is known with atomic precision. As reported recently, researchers have used the cylindrical photosynthetic membrane as a model system to elucidate in great detail how light is captured, and how the light energy is passed around the light-capturing proteins until it is utilized to charge the membrane through electron transfer. The theory of these quantum mechanical processes has been described before (see the April 2010 highlight on Light Capture). More information can be found on our photosynthetic core complex website.
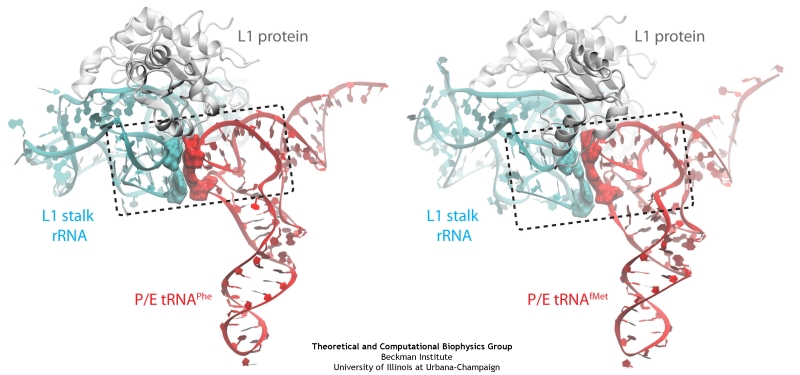
image size:
140.2KB
made with VMD
The ribosome is a molecular machine ubiquitous in all living cells
and translates genetic information into proteins. Proteins are made
of twenty different amino acids, strung in a linear sequence. The
amino acids are coded for by the genes in DNA, but for the purpose of
protein synthesis genes are transcribed into a working copy, a
messenger RNA. The latter is translated by the ribosome into proteins
with the help of transfer RNAs, which bring the individual amino acids.
There is a transfer RNA for each of the twenty amino acids. Much progress has been made regarding the static structure of the ribosome, transfer RNA, and nascent protein components (see also the Dec 2009 and Jan 2009 highlights Managing the Protein Assembly Line and Open Sesame). Now
researchers are looking into the inner workings of the whole system
combining various experiments and computational modeling using
NAMD and
VMD.
The combination yielded unprecedented detailed views of the
ribosome in action as
reported recently,
namely how a dynamic part of the ribosome helps guiding transfer RNAs on
their way out of the ribosome, and explains why transfer RNAs behave
differently on their journey, depending on if they start
synthesis of a protein or if they elongate a protein.
More on our ribosome
website.
Fever, chills, sore throat, coughing, aches, and pains? Ah ..... you have the flu! As a measure of prevention, vaccines against seasonal influenza are distributed and administered each fall. Last year though, the outbreak of the H1N1pdm "swine" influenza virus, caught health workers by surprise as this virus not only infected individuals during the spring and summer months, but also seemed to be particularly virulent towards otherwise healthy young people. Even more alarming was increasing evidence that H1N1pdm had acquired resistance to the frontline antiflu drug, Tamiflu. In response to this, computational biologists at the University of Illinois and the University of Utah teamed up to uncover the basis for influenza drug resistance through quantum chemistry, and molecular dynamics simulations with NAMD. The results of this study have recently been reported, and uncovered a two stage binding pathway for Tamiflu in H1N1pdm "swine" and H5N1 "avian" flu proteins, as well as a possible mechanism through which genetic mutations can induce drug resistance in one of the stages. Subsequent efforts at drug design against influenza can take advantage of this discovery. This discovery was made possible through use of so-called GPU computing (see Oct 2007 highlight "Graphics Processors Speed Up Simulations"). More information can be found here.
Nov ..." />
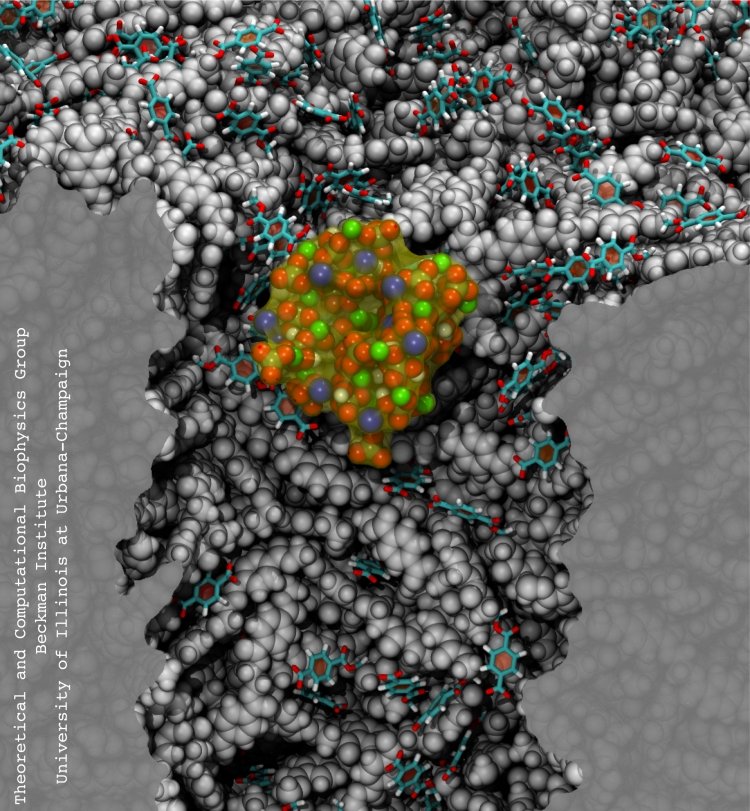
Nanopores are promising new sensors, which may soon sequence DNA in a cheap and fast way (see Nov 2005 and Oct 2004 highlights) and control the flow of ions in nanodevices (see Dec 2009 and Mar 2009 highlights). In measurements, DNA molecules and ions pass through the nanopores which are only a nanometer (0.000001 mm) wide. Unfortunately, researchers have to battle clogging of the very narrow nanopores. For example, it was found that small concentrations of so-called divalent cations produce clogging of plastic nanopores. To figure out how to prevent clogging, researchers needed a microscopic view of the process. Such view was furnished through molecular dynamics simulations, based on NAMD, of ion conducting nanopores. In a recent study, an atomic-level view of the clogging process, revealing the formation of tiny ionic crystals inside the nanopore, was offered. The view suggests a remedy against nanopore clogging. More information can be found in our polymeric nanopore website.
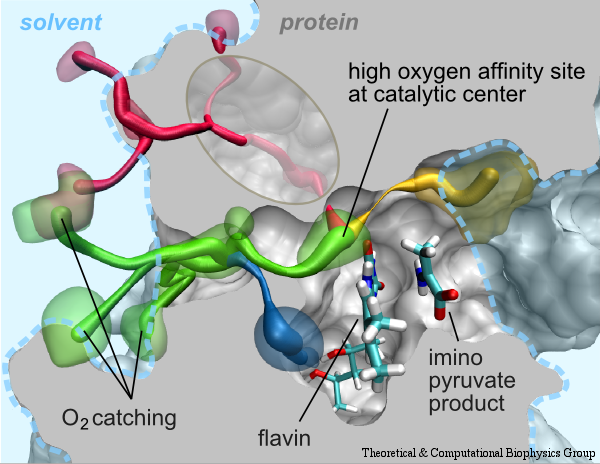
image size:
225.2KB
made with VMD
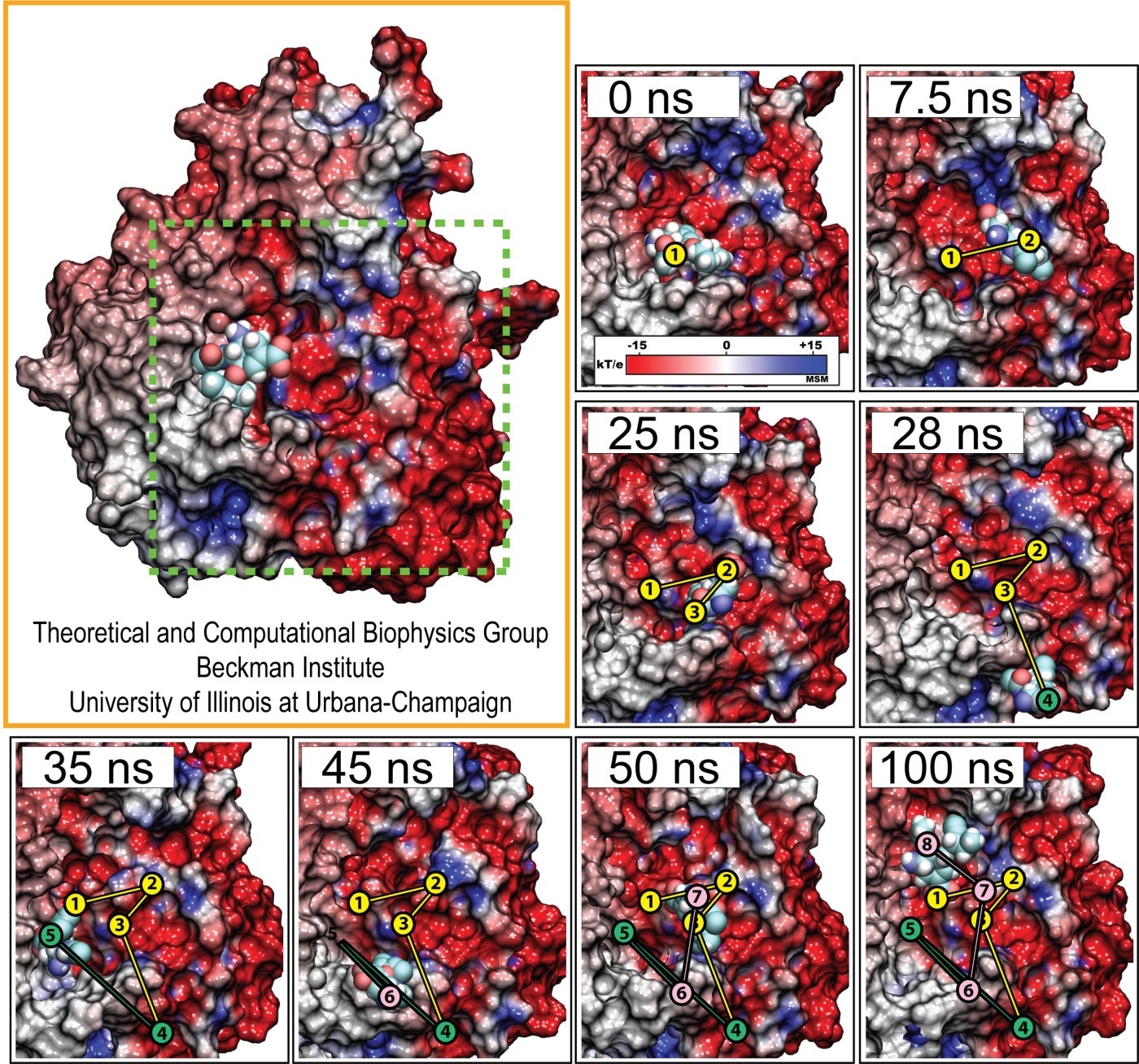
Cells contain numerous enzymes that use molecular oxygen for their reactions. Often, their active sites are buried deeply inside the protein, which raises the question whether there are specific access channels guiding oxygen to the site of catalysis. Localizing oxygen molecules in proteins is difficult. Oxygen is very mobile and difficult to locate by most experimental techniques. However, computer modeling can come to the rescue. In a recent study computational biologists using NAMD could identify the D-amino acid oxidase regions where oxygen molecules most likely reside. D-amino acid oxidase is considered essential for understanding catalysts that involve flavin moieties in proteins. The reported findings reveal a channel system through which oxygen molecules diffuse from the protein surface to the protein's catalytic center. Indeed the channel ends at a site in the protein that constitutes the ideal location for oxygen to react with flavin. The computational results are reported to be verified through so-called site directed mutagenesis experiments. Click here to read more.
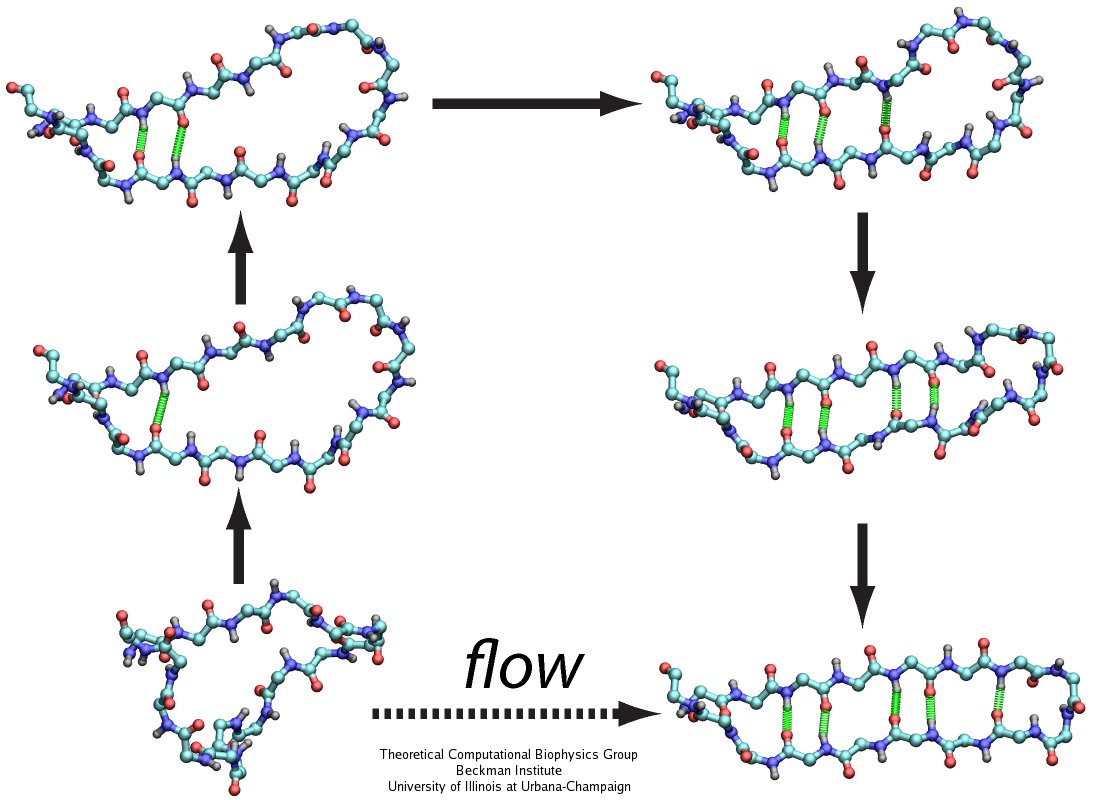
image size:
110.0KB
made with VMD
The human body is protected by self-healing mechanisms, one of them being instant blood clotting at a bleeding site after blood vessel injury. What triggers the formation of a blood clot? Researchers found that a protein on blood platelets, called GPIbα, functions as a sensor of so-called high shear flow caused by bleeding. A loop-shaped, 17-amino-acid-long, segment of GPIb&alpha, the β-switch, acts as the flow sensor. Once a blood vessel is injured, bleeding increases shear stress due to blood flow at the wound, which in turn induces the β-switch to change from a loose, loop-shape to an elongated, hairpin-shape, the latter referred to by researchers as a β-hairpin. This conformational change makes GPIbα stick better to the damaged vessel and eventually leads to blood clotting, which heals the vessel. In a prior study (see the Jul 2008 highlight, Molecular Flow Sensor Triggers Wound Healing), Molecular dynamics simulations using NAMD and VMD provided already a microscopic view of the flow-induced loop to β-hairpin transition. A recent study extended the investigation of the remarkable biological flow sensor, detailing the flow rate needed to trigger it and identifying the detailed sensor mechanism. A combination of simulation and mathematical analysis revealed the β-switch as a system of two stable states, one disordered, with loop geometry and one ordered, with β-hairpin geometry. Normal flow prefers the disordered state; high shear flow prefers the ordered state, inducing thereby the life saving transition. More on our flow sensor website.
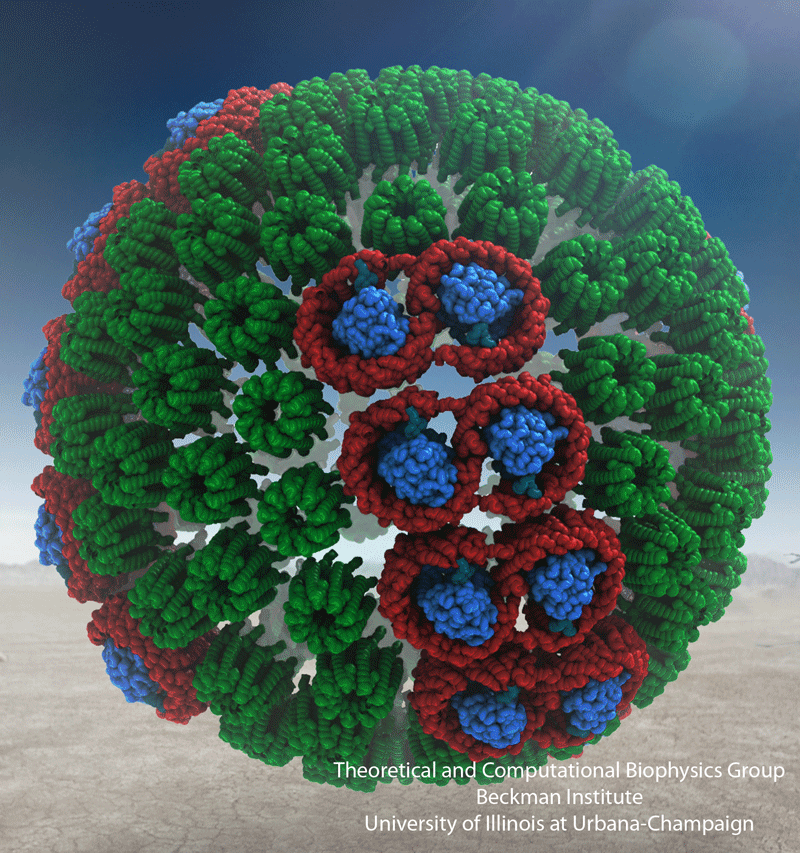
The energy of the sun feeds life on Earth and is harvested by
molecules of chlorophylls and carotenoids. In case of certain
photosynthetic bacteria thousands of these molecules are fitted
into spheres smaller than 1/10000 of a millimeter
(see the October 2007 highlight on
Life's Solar Battery).
Naturally, it is crowded in these spheres!
Researchers now established the composition and arrangement of
the light harvesting system, the chlorophylls and carotenoids
being held by hundreds of ring-shaped proteins. Utilizing the
sun light's energy optimally is a difficult balancing act in
molecular engineering: on the one hand the proteins need to
pack closely to share light energy absorbed by any chlorophyll
or carotenoid, on the other hand, roadways need to be kept open
for passing chemical substances that capture the light energy
in chemical form and deliver it to the remainder of the
bacterial cell. Researchers now report
how the bacterial cell manages the balance through
ingenious utilization of quantum electronics. More on
light-harvesting in purple bacteria can be found here.

credit: Vita Solovyeva
image size:
265.0KB
Animals as varied as salmon, honeybees, and migratory birds have a magnetic sense that sees the Earth's magnetic field and allows them to distinguish North, South, East, and West. In migratory birds the magnetic sense is apparently based on a protein called cryptochrome that acts in the retina, the light-sensitive part of the eyes. In their sensing function cryptochromes rely on light-dependent reactions which depend on the protein's orientation in the geomagnetic field. The product of the reaction in cryptochrome is said to affect the sensitivity of light receptors in the retina of a bird's eye such that a bird literally "sees" the geomagnetic field. However, cryptochrome in the eye most likely is not perfectly aligned with the retina, even if bound to ordered membrane structures found in the outer segments of the eye's light receptors. In a recent report, researchers have shown that birds' vision-based compass is surprisingly insensitive to cryptochrome disorder. The researchers suggest a cloud-like pattern in the visual field that points a bird to the correct orientation. More on our cryptochrome web site.
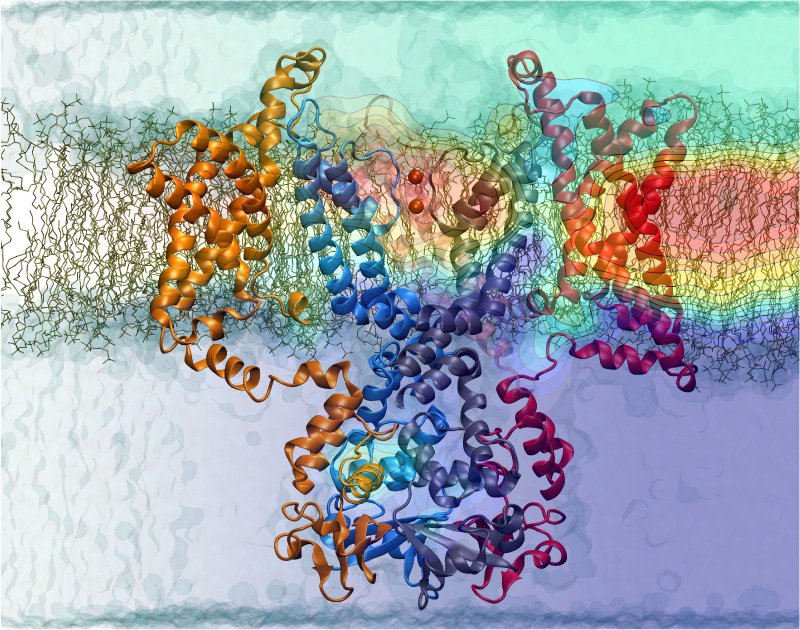
Nerve cells, through their electrical signals, control actions and intelligence of higher organisms. The signals result mainly from potassium and sodium ion channels in the cells: when the cells are stimulated electrically, they send an all (in case of sufficient stimulation) or nothing signal to other nerve cells or organs like muscle. As shown in ground breaking work by 1963 Nobelists Hodgkin and Huxley, cast into mathematical equations, nerve cells establish these signals through voltage gating of channels. The nature of the gating, monitored through the so-called gating current, has been elusive for decades, despite a detailed characterization of the ion conductivity itself rewarded through a 2003 Nobel Prize to MacKinnon. The riddle is that the channel involves a protein with few charged amino acids that seem to be only weakly coupled energetically to an electrical potential gradient across the cell membrane. Now a sweeping modeling study using NAMD employing the most powerful computers available to researchers today has led to an explanation of voltage-gating. Simulations revealed that the potential gradient is focused by the channel protein to a very narrow region such that its value is much larger than anticipated. The protein was also seen to arrange its charged amino acids sensing the gradient in an unusual helix, a so-called 310 helix, that aligns charges perfectly while at the same time inducing a motion that opens and closes the channel. Proof of the veracity of the computational model is that the calculated gating current fits perfectly the observation. More on our potassium channel website.
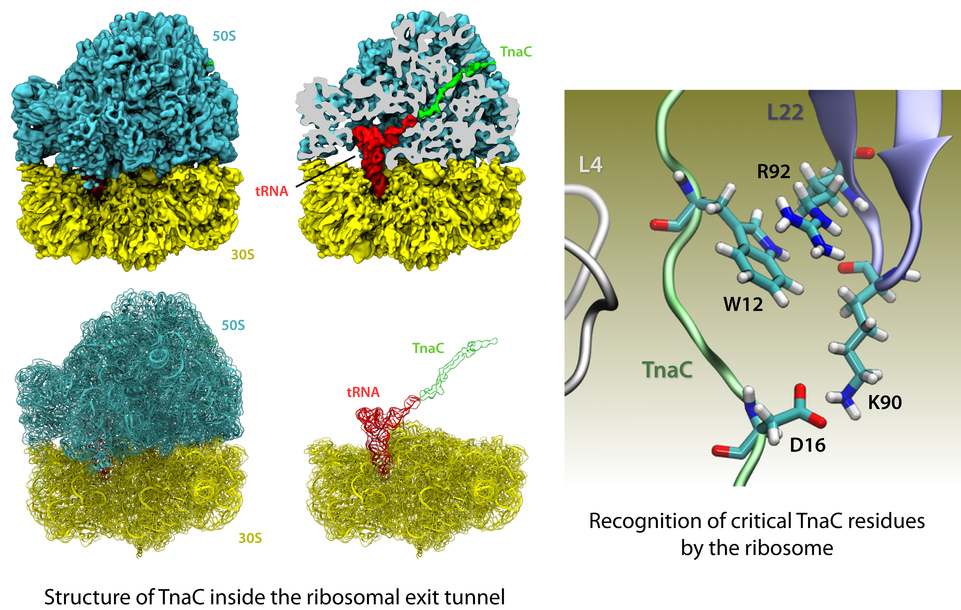
image size: 541.6KB
The ribosome is the protein factory of all cells. While proteins are
being synthesized, they must travel through a tunnel inside the
ribosome before they reach their required location in the cell. This
tunnel recognizes certain sequences of nascent proteins and responds
to them in various ways. For example, certain protein sequences
recognized in the tunnel can shut the ribosome down, stopping protein
synthesis. A much studied example is the bacterial protein TnaC, which
by shutting down its own synthesis turns on the synthesis of proteins
involved in the degradation of a molecule called tryptophan. The
structure of TnaC inside the ribosome was previously determined through
crystallography, electron microscopy, and computer modeling (see the December 2009 highlight on
Managing the Protein Assembly Line). Simulation of TnaC dynamics inside the ribosome using
NAMD and
VMD has now revealed the mechanism by which TnaC shuts down
the ribosome as reported recently. Answered was also the
question of how the ribosome recognizes the sequence of TnaC and why many
proteins that have sequences similar, but not identical, to that of
TnaC do not shut down the ribosome. For more details, see our
ribosome website.
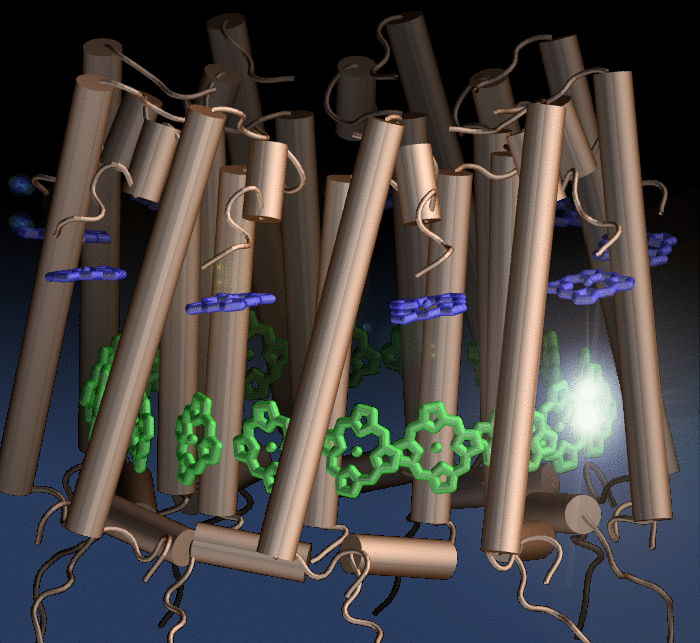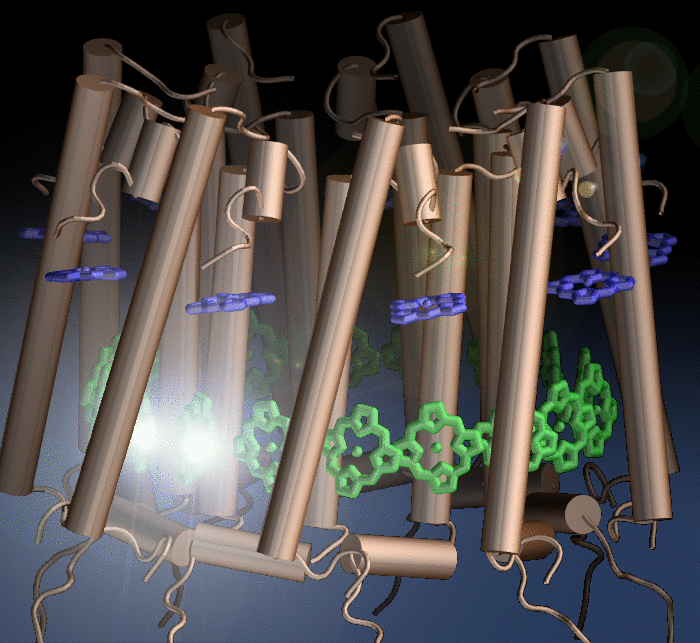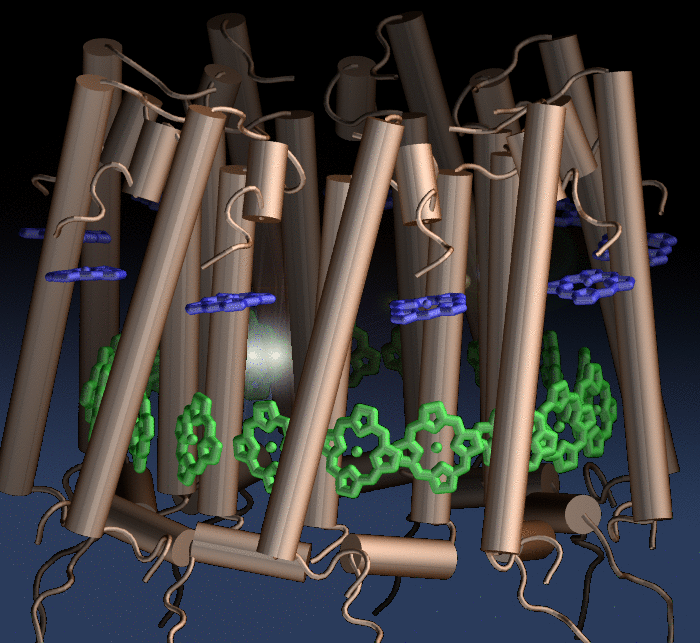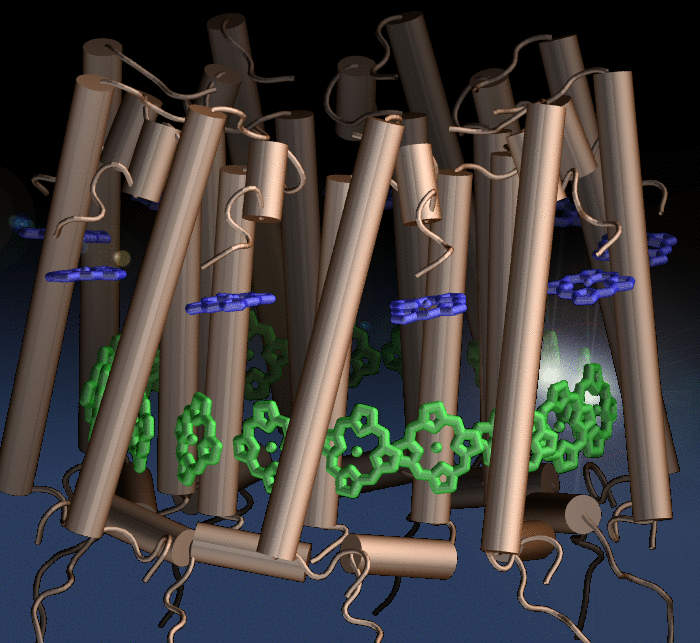
Photosynthetic organisms fuel their energy needs by harvesting sunlight. Photosynthetic bacteria called purple bacteria live in the darkest places in ponds and lakes and as a result, every photon of sunlight that they receive must be efficiently collected and converted into chemical energy for the cell. Incoming photons are absorbed by proteins that contain many molecules of chlorophyll (see the October 2007 highlight on Life's
Solar Battery). The chlorophylls capture light energy and transfer that energy between them. Such transfer, a quantum mechanical process, happens multiple times before the energy is converted to chemical energy and is governed by thermal interactions between chlorophylls and protein. A recent report showed that the surrounds thermalize, i.e., randomize, the light excitation migration within one picosecond (0.000000000001 second), after which the typically coherent quantum process looks incoherent. The finding explains the great success of earlier simpler descriptions suggested sixty years ago by the US physicist Robert Oppenheimer and German chemist Theodor Förster who postulated the now established incoherence. More here.
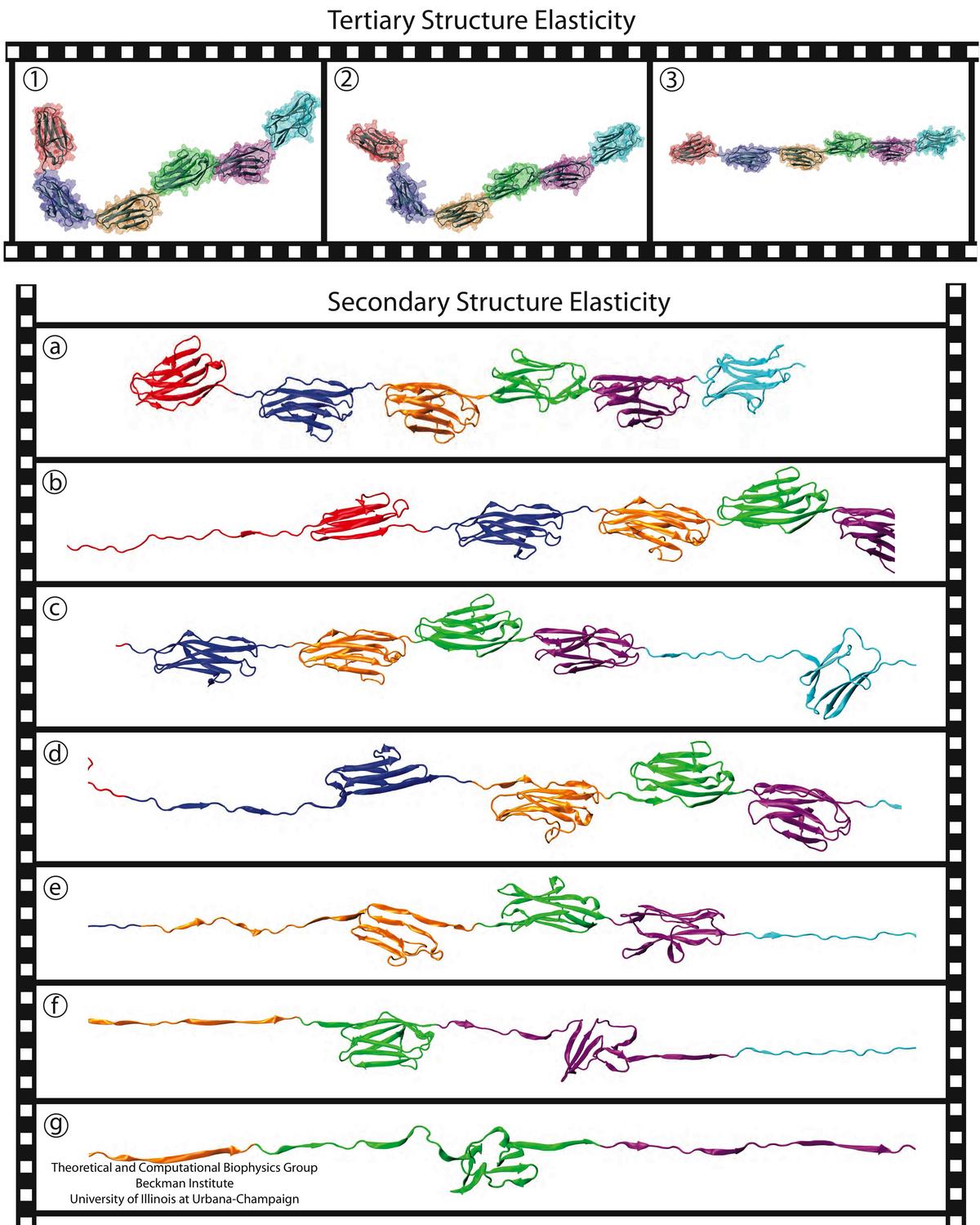
image size:
212.3KB
made with VMD
A smart strategy usually involves a plan B. As it turns out, the muscle proteins in our bodies responsible for the physical motions like running or the beating of our hearts, also rely in their function on having a plan B strategy. When contracting and extending, muscle fibers generate tremendous forces that need to be buffered to protect muscle from damage. This role falls to the muscle protein titin, which is composed of a chain of linked domains, making it a molecular rubber band. When a small force is applied, titin employs its plan A and stretches apart without unraveling its individual domains (like what the movie on the side shows). When a stronger force is applied, plan B kicks in and further elasticity is generated by the unwinding of the protein domains one at a time. By practicing two modes of response to different levels of forces, titin provides the elasticity that muscle needs at a minimal structural cost. A recent computational-theoretical investigation has provided a molecular view on how titin's two plans work, the study featured in a journal cover. The needed simulations were performed using NAMD. Principles described in this study can also be found in other mechanical proteins, recently reviewed here. More on our titin IG6 website.
image size:
180.0KB
made with VMD
As the size and complexity of structurally resolved biomolecules
continues to increase, researchers require ever more powerful
visualization and analysis tools to gain understanding of
their function and dynamics.
The latest release of VMD,
the leading molecular dynamics visualization tool, has unleashed the
computational power of
graphics processing units (GPUs)
and multi-core CPUs enabling researchers to interactively study
large biomolecular complexes on desktop computers as a matter of
daily routine.
VMD 1.8.7
includes many new and improved graphics algorithms enabling fast
rendering of high resolution photorealistic molecular scenes.
New graphical representations and coloring features enable
visualization of carbohydrates, nanodevice structures, and
results of quantum chemistry simulations.
Fast GPU electrostatics algorithms reduce or eliminate the need for
batch mode calculations, e.g., for computing electrostatics of
large systems, accelerating them by a factor of 22-44.
The latest improvements to the
MultiSeq
plugin enable processing of up to 100,000 sequences on a desktop computer.
New and updated structure building tools ease construction of
large all-atom and coarse-grained molecular models.
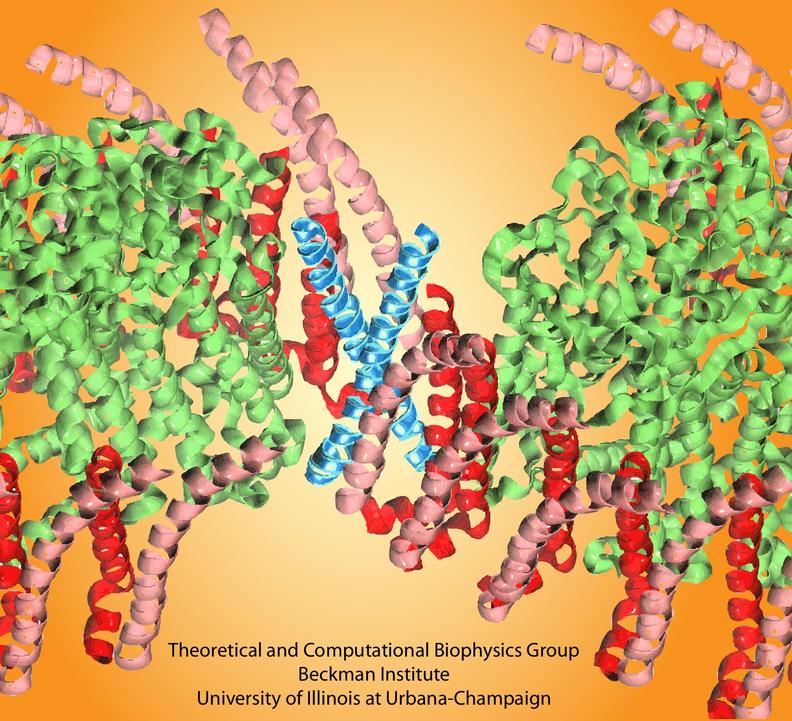
image size:
115.2KB
made with VMD
Bacteria contain the simplest photosynthetic machineries found in nature. Higher organisms like algae and plants practice photosynthesis in a more elaborate but principally similar manner as bacteria. But even for its simplicity, the bacterial photosynthetic unit is not without its unsolved mysteries. Take, for example, the crucial photosynthetic core complex, which performs light absorption and the initial processing of the light energy. In certain bacterial species, the core complex contains two copies of an additional small protein (made of about 80 amino acids) called PufX, whose role in photosynthesis is still a puzzle, and its location within the core complex is yet to be pinpointed. Numerous imaging studies have been published, yielding two opinions on what the role of PufX is and where exactly it resides. One opinion assigns the protein the role of gate keeper, the other the role of coordinator. Recently, a computational investigation was carried out that much supports the second role. Since PufX comes as a pair, two copies of PufX were placed side-by-side in a biological membrane and they were seen to adhere to each other strongly, but assume with their cylindrical (helical) shape an angle of 38 degrees. This geometry is perfectly suited for PufX to join the two parts of the symmetrical core complex together in the middle and to impose on the parts the tilt that was actually observed in the imaging studies. The needed simulations were done with NAMD. More details can be found on our photosynthetic core complex website.



{kind=link}