Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
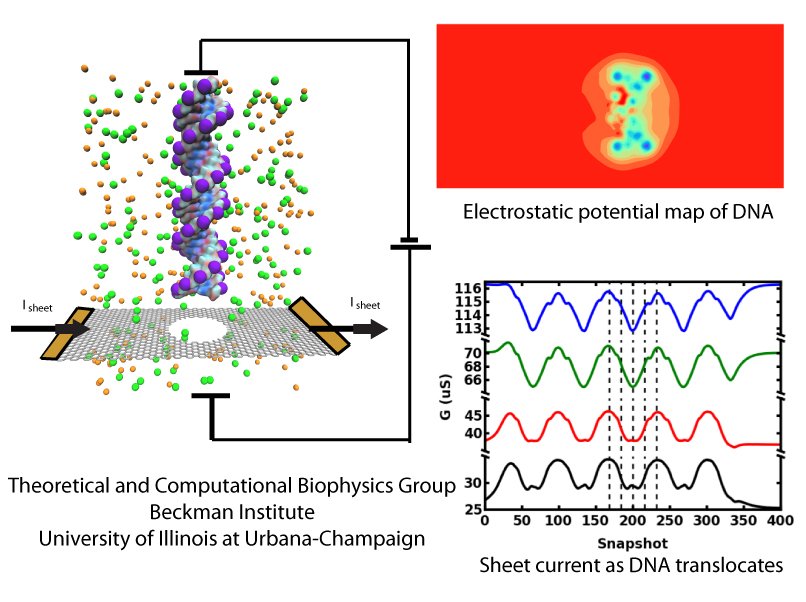
image size:
210.7KB
made with VMD
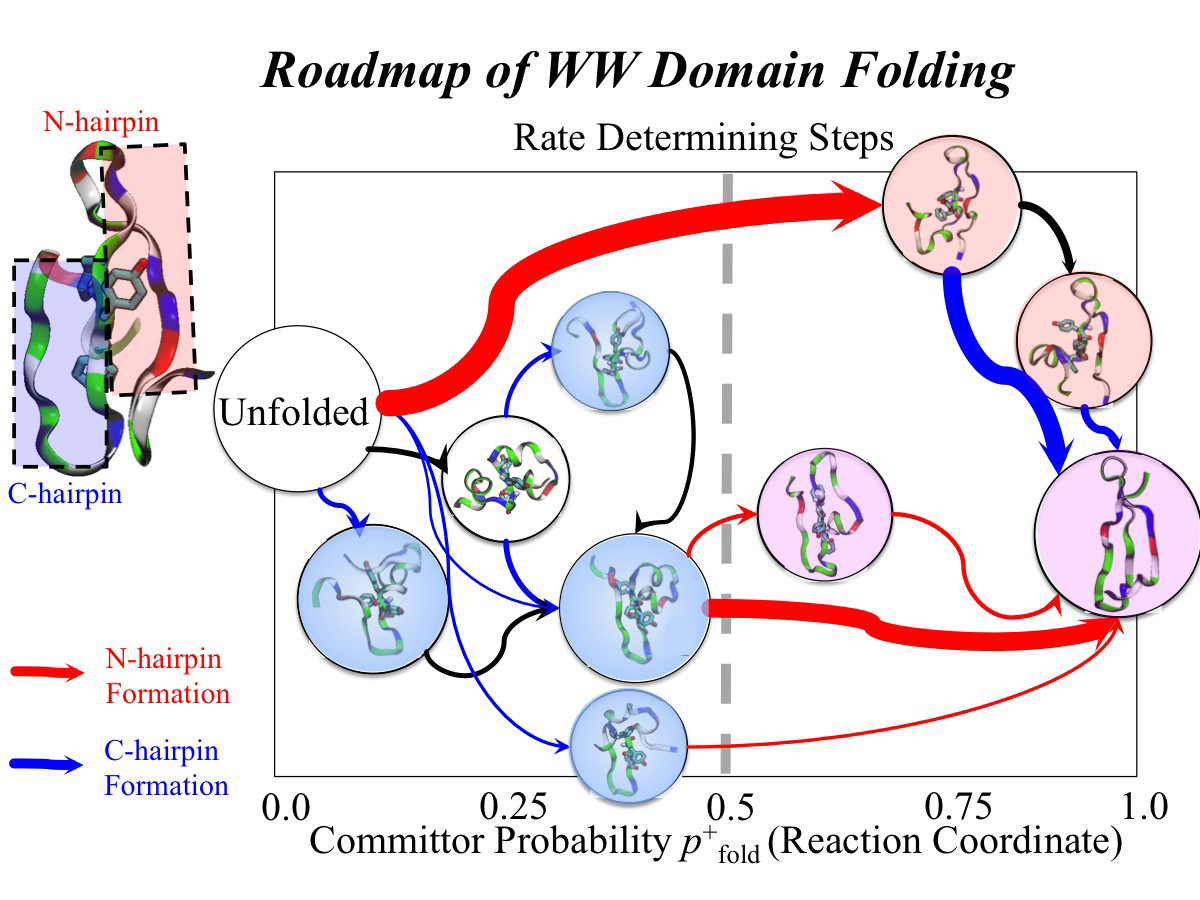
image size:
507.8KB
made with VMD
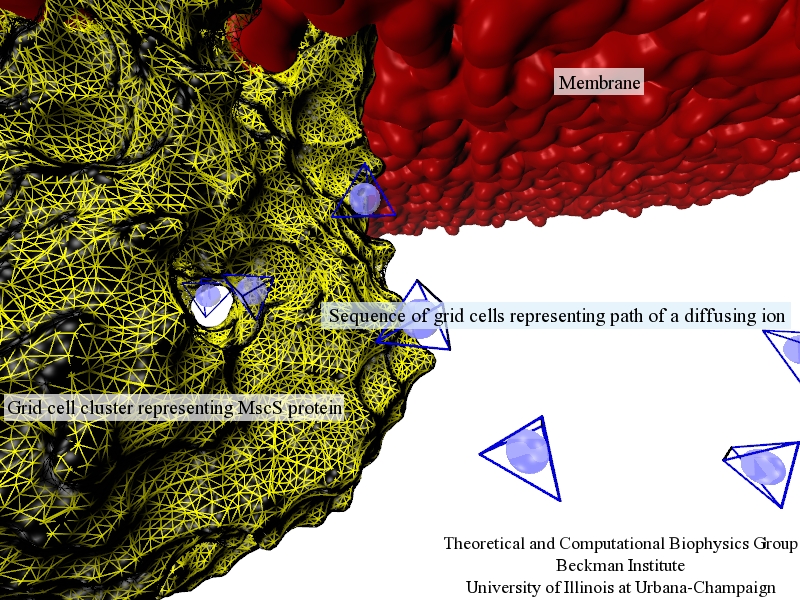
image size:
411.0KB
Traffic flow in a city is affected by large-scale features, like the layout of road networks, as much as it is by small-scale ones, like traffic lights at a road junction. Likewise, the transport of small molecules in cells occur on multiple scales. For example, ions diffusing through the mechanosensitive channel of small conductance (MscS) (see highlights from Jul 2011, "Smart Bacterial Safety Valve", Mar 2008, "Observation and Simulation depict Cell's Safety Valve", Feb 2007, "Observing and Modeling a crucial Membrane Channel", May 2006, "Electrical Safety Valve", and Nov 2004, "Japanese Lantern Protein") must navigate the intricate geometry of the MscS, which varies by the Ångstrom. At the same time, the distribution of ions within hundreds of Ångstroms of the MscS fluctuate as ions escape through the channel, thus changing the electrostatic landscape seen by other ions as they approach the MscS. In order to model both the fine and bulk aspects of diffusion in systems like that of the MscS, scientists have proposed, in a recent report, a method that marries the high spatial resolution of molecular dynamics to long range diffusion. In the new description, biomolecules "diffuse" by hopping through a grid under the influence of Coulomb and other forces. More on our Kinetic Diffusion web site.
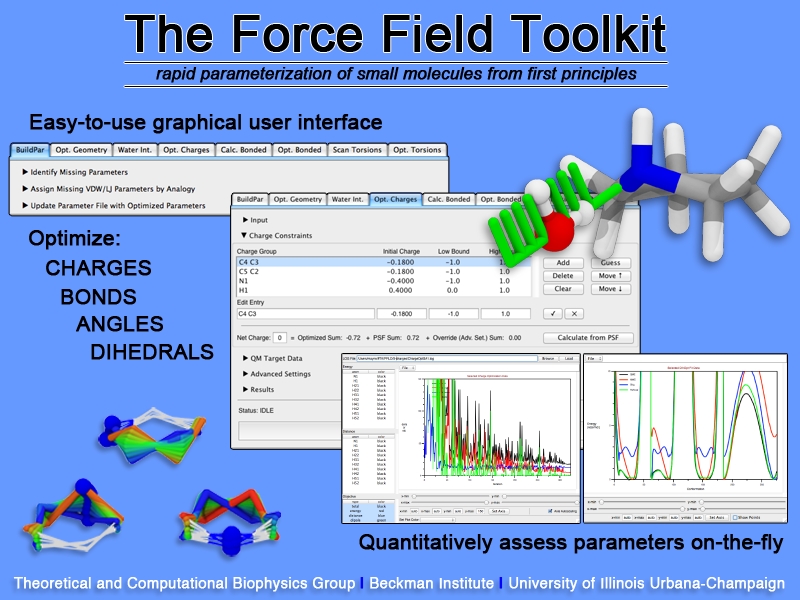
image size:
307.6KB
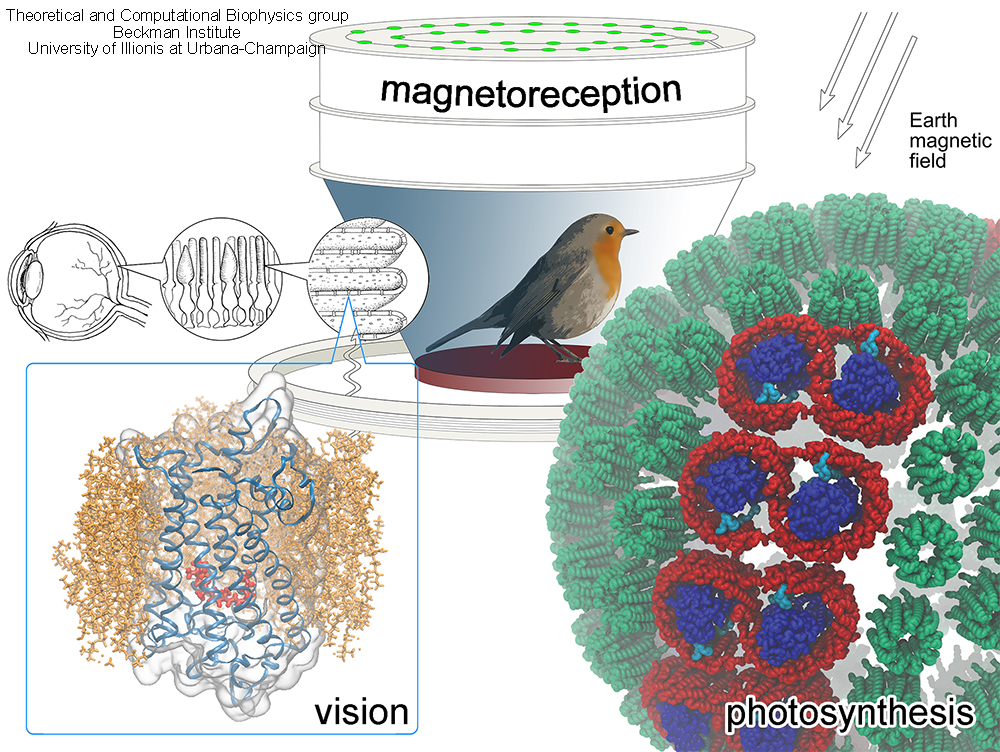
image size:
679.7KB
credit: Olga Svinarski
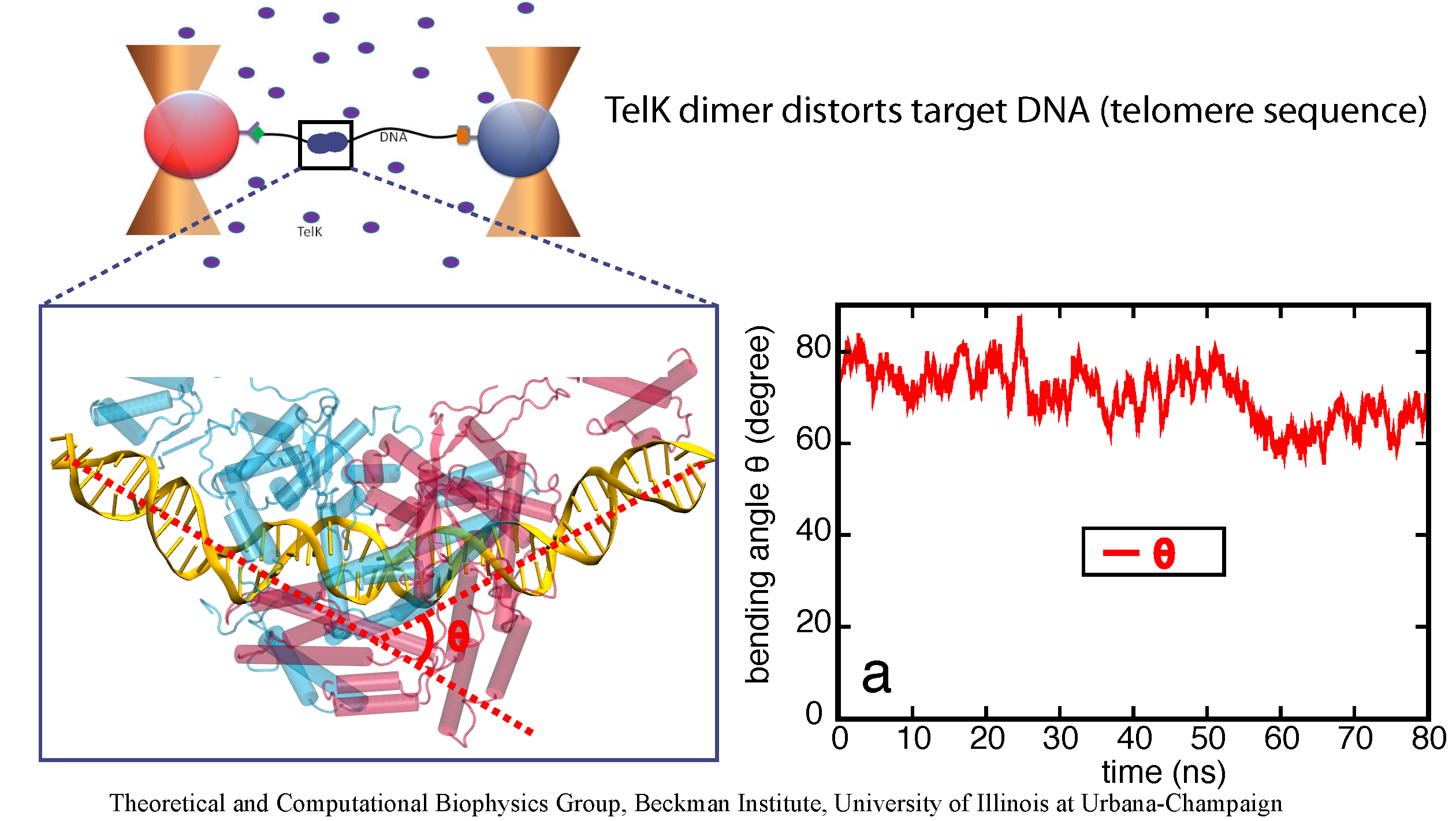
image size:
651.0KB
made with VMD
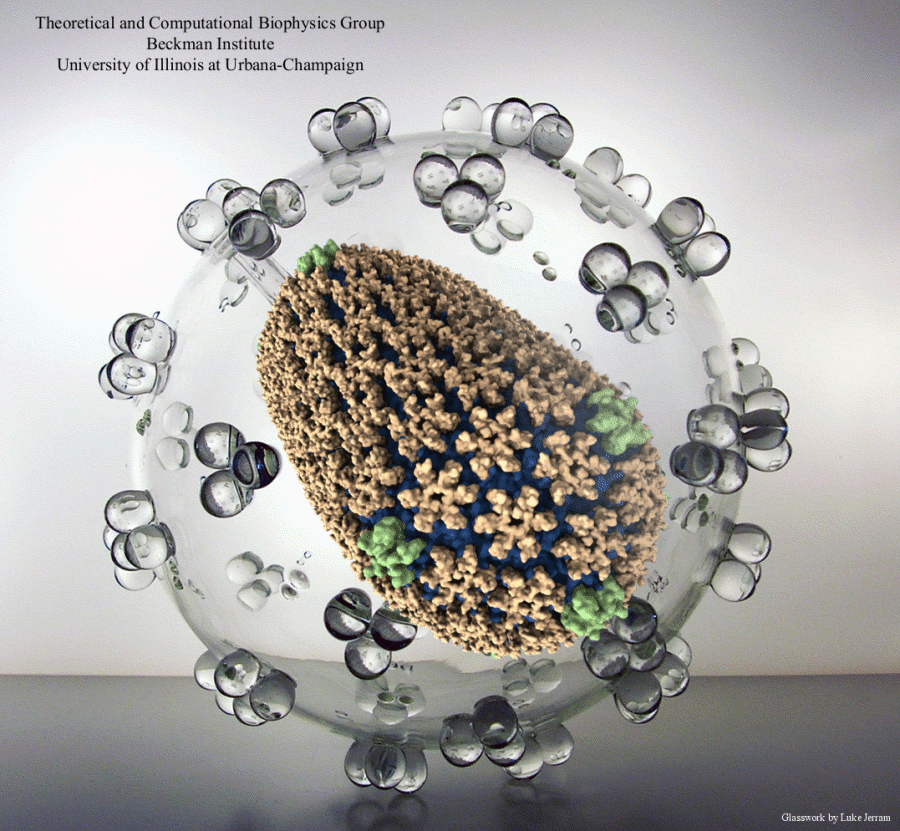
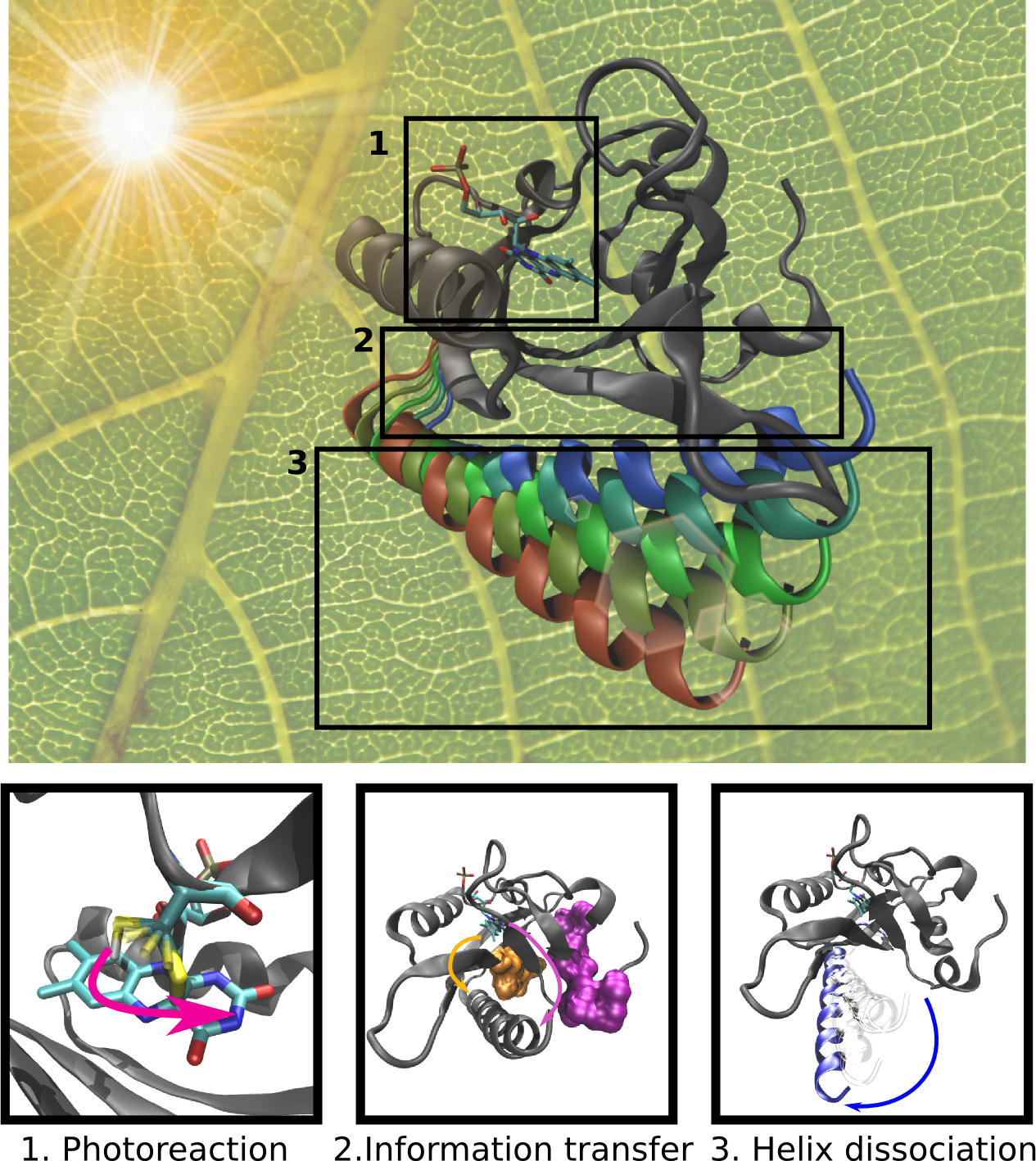
image size:
1.9MB
made with VMD

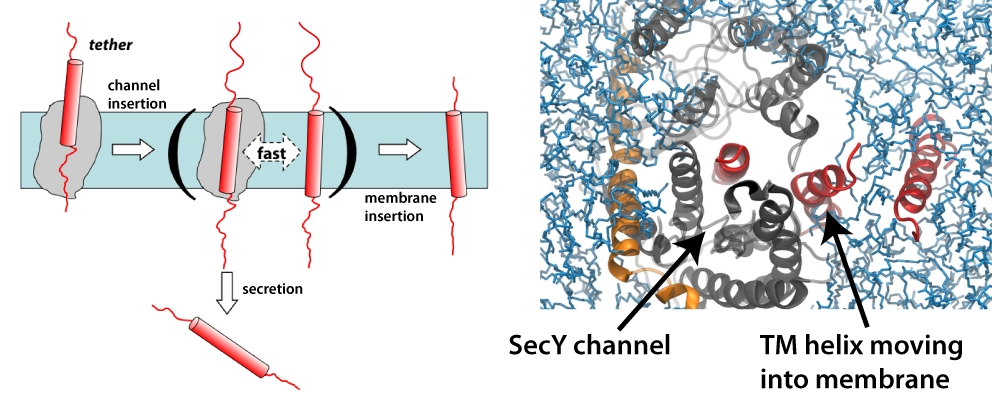
image size:
223.2KB
made with VMD
image size: 1005.1KB
image size:
1.3MB
Olga Svinarski and VMD
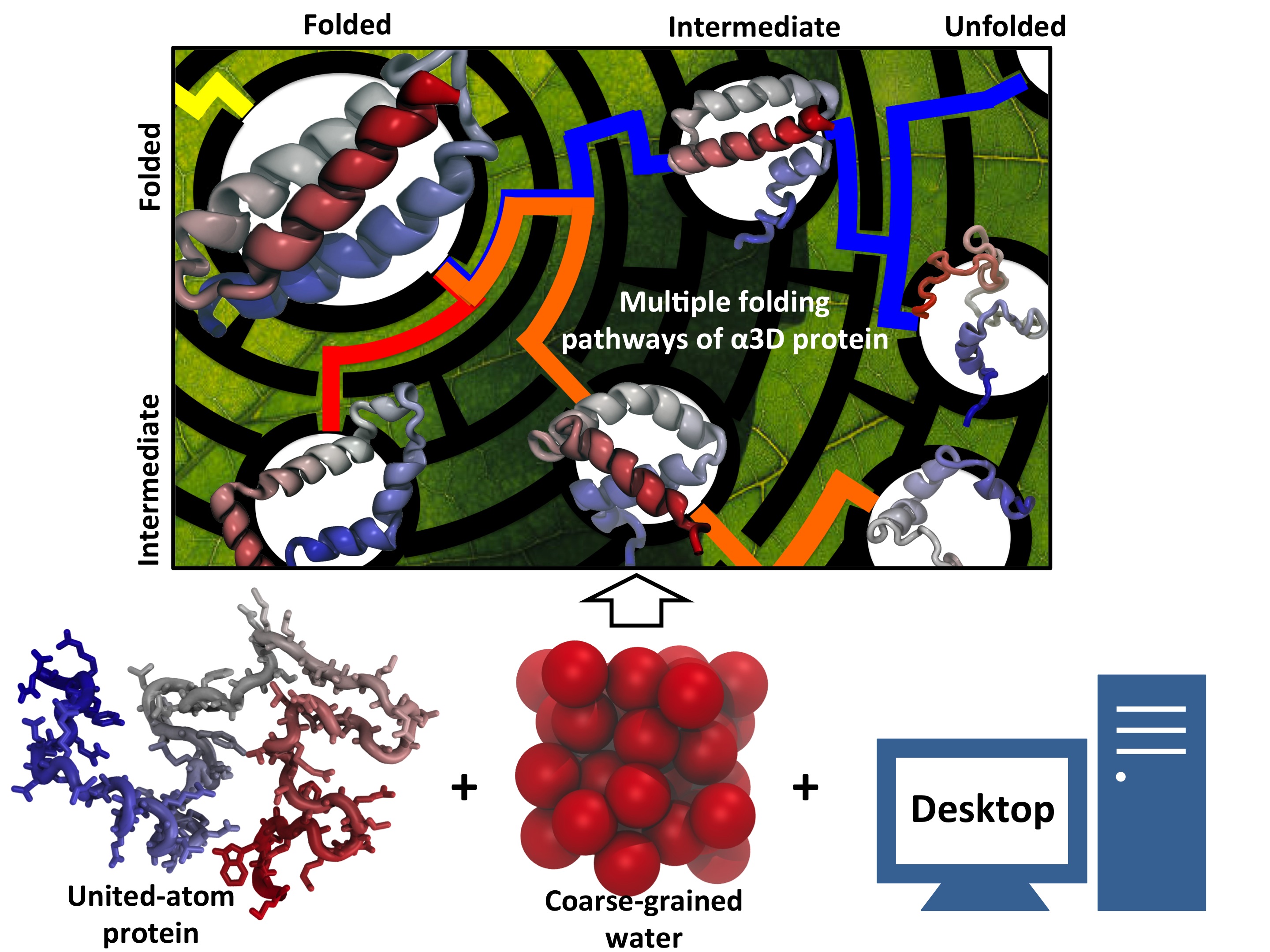
image size:
90.9KB


