Highlights of our Work
2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
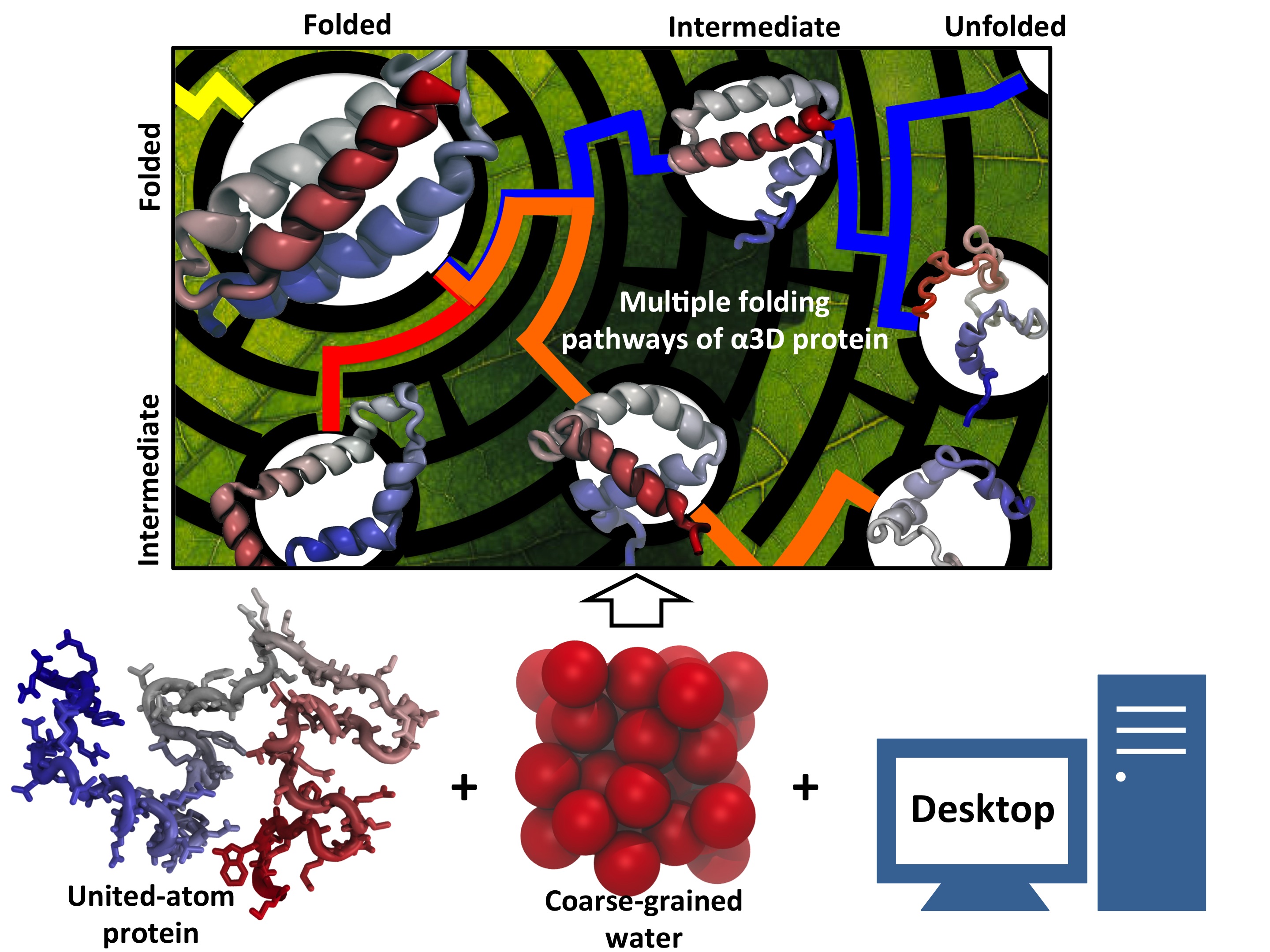
Every living cell relies on proteins to carry out its functional tasks;
every protein needs to assume a proper shape in order to be operational for these tasks. How a protein,
composed of a particular sequence of amino acids,
could find its way to a proper shape is a fundamental, yet mysterious biological process.
Researchers have sought to unravel atomistic details of protein folding processes through computer simulations,
but modeling such processes is computationally demanding.
It was only recently that some researchers have been able to observe in some case
how proteins fold, but needed for the purpose the fastest computers available today.
One of these computers is Anton,
the expensive special-purpose supercomputer available essentially only to a single research group.
Is there an affordable way to simulate protein folding?
One solution could be coarse-grained methods.
These methods save tremendous computational effort by replacing
computational models that include all atomistic detail.
However,
the simplified models need to include a sufficiently accurate description of proteins
for modeling folding processes. As reported recently,
researchers have overcome the challenge by combining atomistic and coarse-grained descriptions.
The new method is fast enough to follow movements of proteins long enough to see them fold,
while requiring only readily available computer powers. The new method allowed
researchers to analyze complete folding events for seven proteins,
including a protein, called α3D (see movie, 11.5 M),
that is one of the largest proteins ever folded computationally.
More on our hybrid-resolution model website.


