Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
~ ~
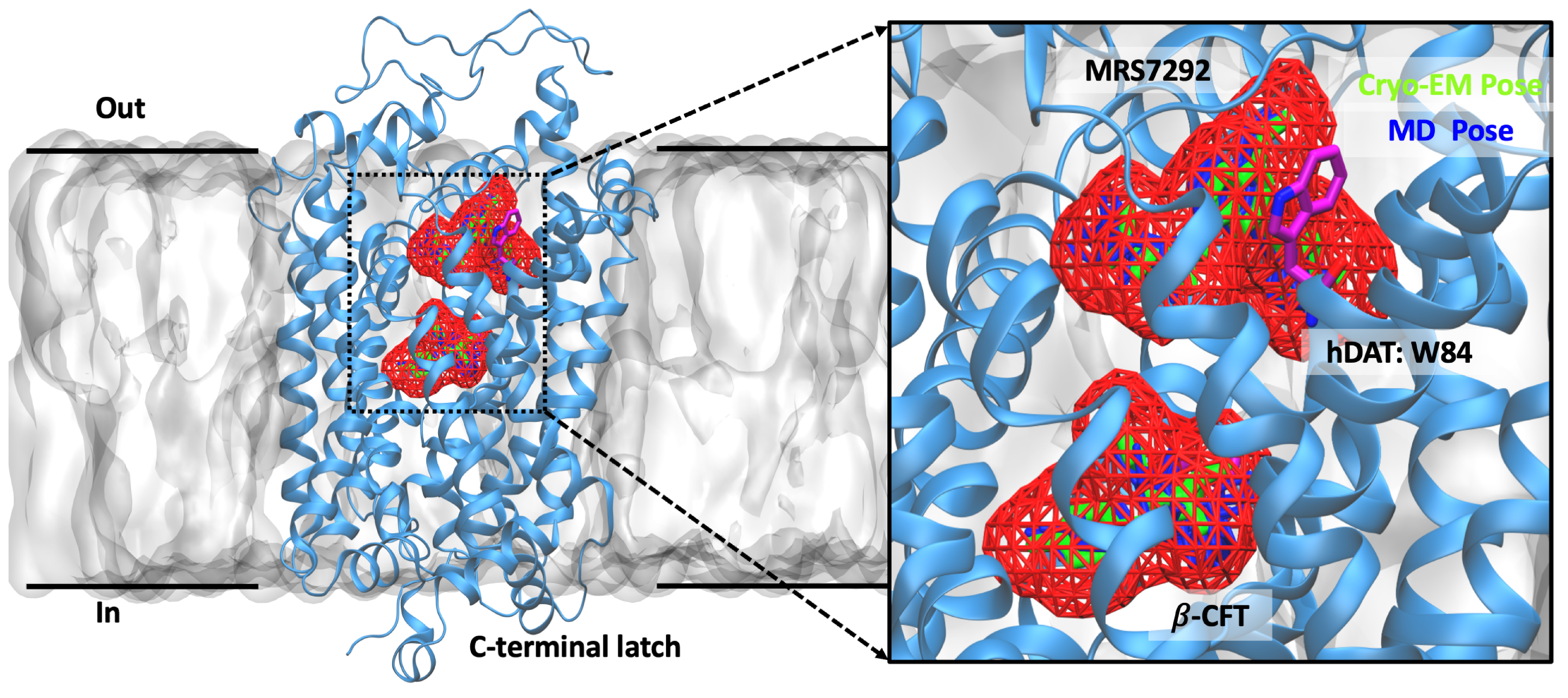
Dopamine is a key neurotransmitter in the brain involved in many processes, including motor control, reward, and cognition. The dopamine transporter (DAT) is a membrane protein responsible for the uptake of dopamine, thus terminating its function. DAT is a key target for drugs treating Parkinson, ADHD, mood disorders, and schizophrenia, and inhibition of DAT is a central mechanism for drugs such as cocaine. Our collaborators in the Gouaux lab at OHSU have captured the structure of human DAT using cryoEM, together with a competitive inhibitor at its central binding site, and an allosteric inhibitor in an external site. However, the precise binding poses and stability of these inhibitors were unclear. To address these issues, the Resource researchers applied molecular dynamics simulations with NAMD/AMBER and analyzed by VMD to explore the most likely binding poses and stability of the inhibitors. For more details, see our recent publication in Nature.

Infections by Gram-negative pathogens are increasingly prevalent and consistently lead the top threat lists of the World Health Organization (WHO) and the Centers for Disease Control and Prevention (CDC). These infections are typically treated with broad-spectrum antibiotics, resulting in widespread disruption of the gut microbiome and increased susceptibility to secondary infections. Recently, our collaborators in the Hergenrother lab discovered a novel antibiotic called lolamicin, which is active against more than 130 multidrug-resistant bacterial species. Notably, lolamicin spares the gut microbiome, preventing secondary infections.
As highlighted in a recent publication in Nature, Resource researchers used molecular simulation with NAMD 3.0 to characterize the binding of lolamicin to its target, an ABC transporter known as LolCDE. By identifying the major binding pose and intermediate poses, we gained insights into how this compound engages its target and how to design next-generation lolamicin derivatives.

The recent pandemic has underscored the critical importance of understanding viral structure and function to prevent their spread. By employing a multiresolution simulation approach that utilizes ARBD and NAMD 3.0, researchers at the Resource revealed how a DNA virus organizes its genome through repeated tight folding of the DNA rather than the traditional view often depicted in textbooks. Atomistic simulations further confirmed consistency with available experimental data, including pressure and average structure. These groundbreaking findings were recently published in Nature.

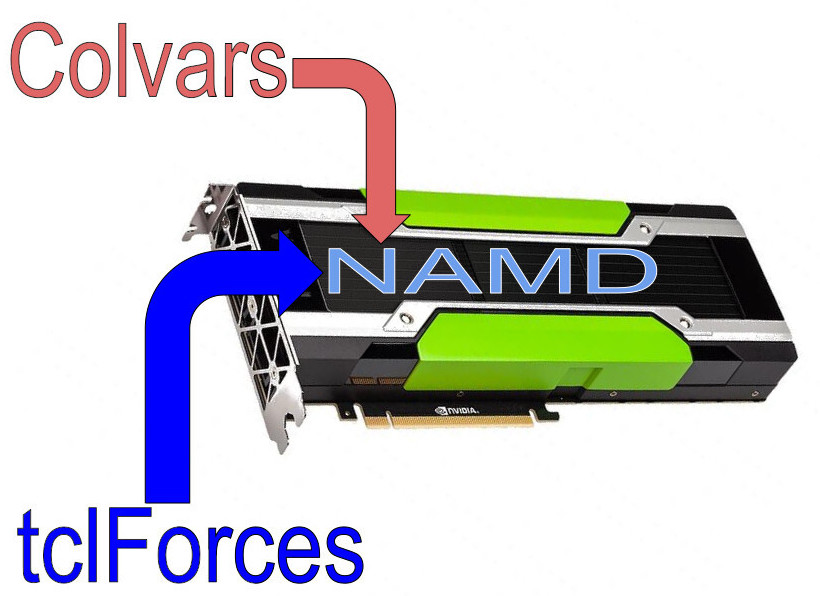
We are excited to announce the availability of the AMBER force field in VMD/NAMD, enhancing our ability to prepare and run molecular dynamics (MD) simulations with different force fields. Utilizing the PSFGEN file builder, available as a VMD plugin, one can now apply the AMBER force field natively in VMD 1.9.4 without tedious constraints file formatting. This flexibility allows for the study of large systems that were previously impractical to model with AMBER alone. The integration leverages the strengths of AMBER, renowned for its precision in modeling biomolecular systems, and brings it into the VMD and NAMD environment, known for their ability to run large systems efficiently and for their powerful visualization and analysis capabilities. Researchers can now explore complex biomolecular dynamics with unprecedented accuracy and scale, opening new avenues for groundbreaking discoveries in structural biology. More details about AMBER for NAMD can be found in a recent publication in the Journal of Chemical Information and Modeling.
With the continued global warming, carbon capture has turned into a highly relevant subject to our daily lives. To discover optimal materials for this purpose, we have turned to an innovative method integrating artificial intelligence (AI) and molecular simulations. Leveraging AI and machine learning, over 120,000 new candidates were generated within minutes. High-throughput screening and molecular dynamics simulations were then used to evaluate their stability and carbon capture capacity. As highlighted in a recent publication in Nature Communications, this innovative approach holds potential not only for advancing carbon capture technologies but also for addressing broader challenges in biomolecular simulations and drug design.
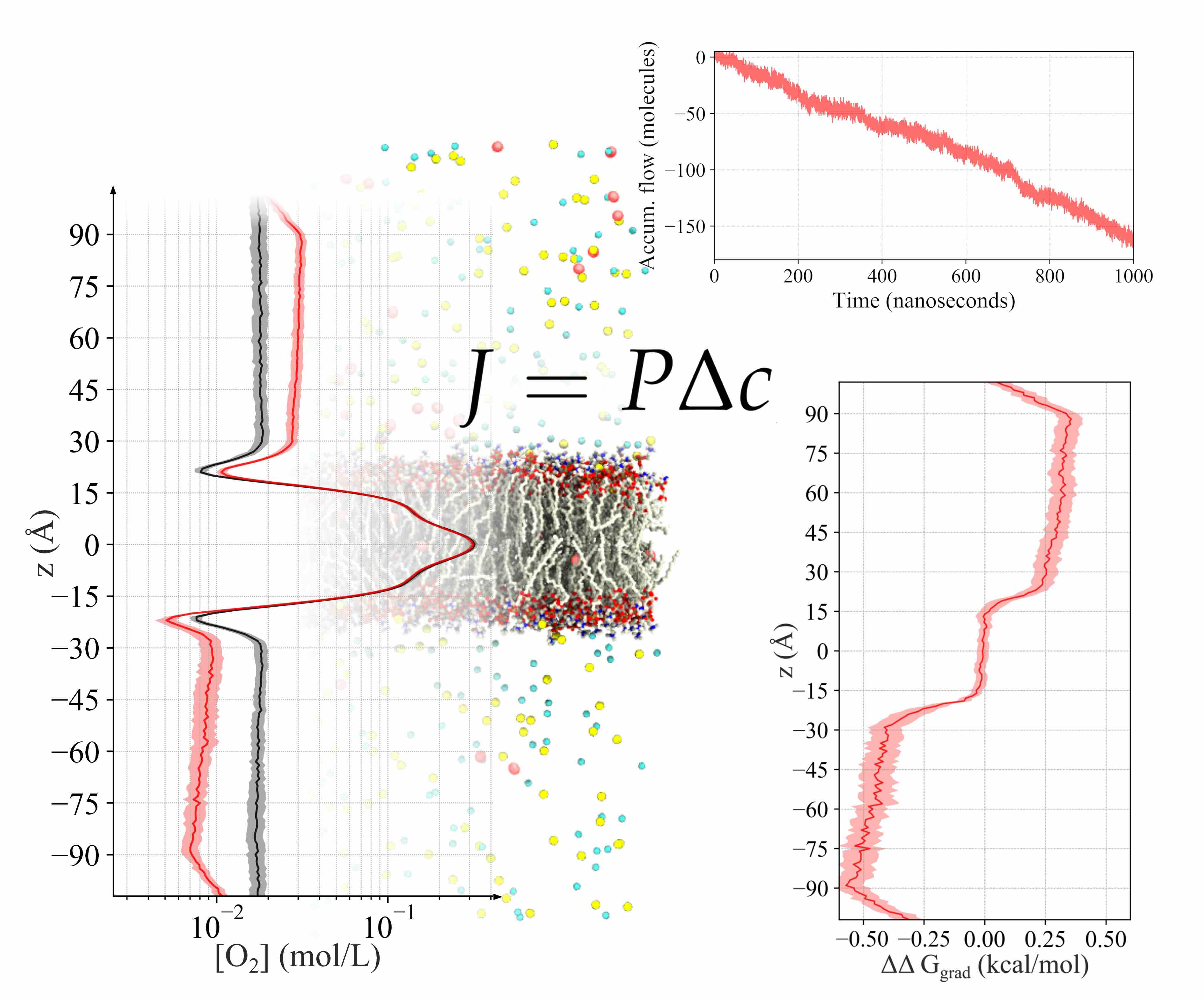
Permeation of metabolic substrates across biological membranes is a fundamental process in cellular life. This process is largely driven by the concentration gradient of various molecules between the outside and inside of a cell. To meet the need for creating such concentration gradients in MD simulation, and to calculate permeation under natural conditions, we developed a technique in NAMD to continually drive permeant molecules near the periphery of the simulation box across the periodic boundary, which results in a sustained gradient in the center of the simulation system where the membrane is located. This allows for purely diffusive motion of particles across a membrane, enabling one to directly calculate permeability the same way as in experiment. Read more in a recent paper.