Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
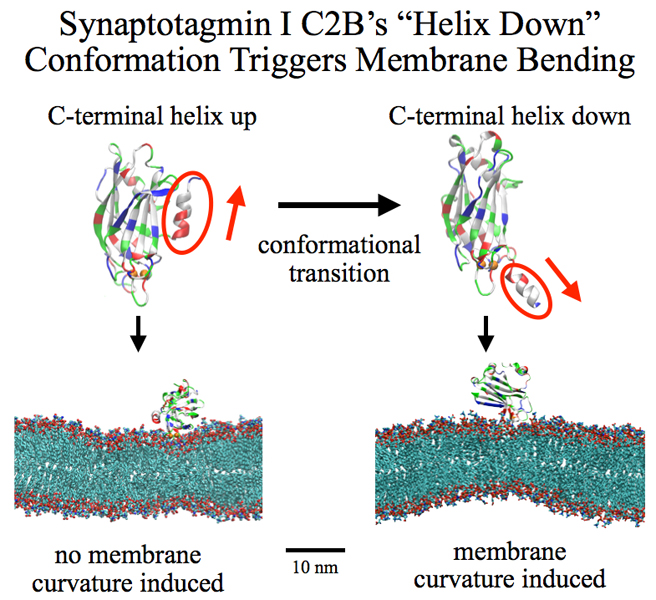
image size:
258.8KB
made with VMD
Neurons in the brain form a closely knit communication network with other
neurons. Each neuron sends messages through up to thousands of cell-cell communication
channels, so-called synapses. To avoid communication chaos, the messages of each neuron
are chemically encoded as if neurons speak English to some neurons and French
to others. The neurons employ an extremely efficient
encoding system, packaging chemical message molecules, so-called neurotransmitters,
in spherical vesicles encapsulated by a lipid membrane just like the whole neuron is
encapsulated by a membrane. The vesicles aggregate near the presynaptic site of the membrane,
ready to release their neurotransmitters into the space between neurons at the synapse,
the so-called synaptic cleft. When a sender neuron becomes electrically active, as it
wants to "speak",
the electrical activity releases
Calcium ions at the pre-synaptic cell that trigger merging
(fusion) of vesicles with
the sender neuron's membrane.
At this point the neurotransmitter molecules flow into the synaptic cleft.
The receiver neuron "hears" the signal by receiving the neurotransmitter
molecules on receptors in the postsynaptic membrane, inducing as a result an electrical
signal in the receiver neuron.
The release
involves a group of proteins that make vesicles ready for the release and
proteins that execute the Calcium-triggered step, among the latter
synaptotagmin I. As reported recently, researchers have proposed
with the help of computer simulations using NAMD how
synaptotagmin I acts.
The simulations suggest that the experimentally observed structure of synaptotagmin
I measured in vitro in a crystal/NMR form of the protein differs from the
active, in situ form of synaptotagmin I.
The finding, if true, will be a dramatic example for the role of computing in biology
where the computer often complements observation studying, as in the present case,
biomolecules in situ, namely their natural environment, rather than in vitro,
namely in an artificial environment.
Please read more on our neuron transmission website.

image size:
63.5KB
see also screencast
Who needs a supercomputer to do molecular dynamics? Thanks to Theodore Gray's new "Molecules App" for iPad/iPhone, you can now do molecular dynamics literally with the touch of a finger. "Molecules" by Theodore Gray is the interactive version of his beautifully illustrated book on the properties of the molecules that make up our world. Integrated into "Molecules" is the molecular dynamics code NAMD, which makes it possible to run real-time interactive simulations of the molecules in the book. You can pull, stretch, and twist hundreds of different molecules, or even tie them in knots! By playing with the interactive molecular dynamics simulations, readers can get an intuitive feel for the properties of molecules - not to mention, they are just plain fun to play with. The "Molecules App" is already very popular, and has been chosen for the Editors' Choice list on the App Store. You can see the app for yourself in the iTunes App Store, read more about the creation of the app on Theodore Gray's blog, and see a screencast of honorary group member Sebastian (pictured here) playing with NAMD through the app on the 500 atom molecule maitotoxin.
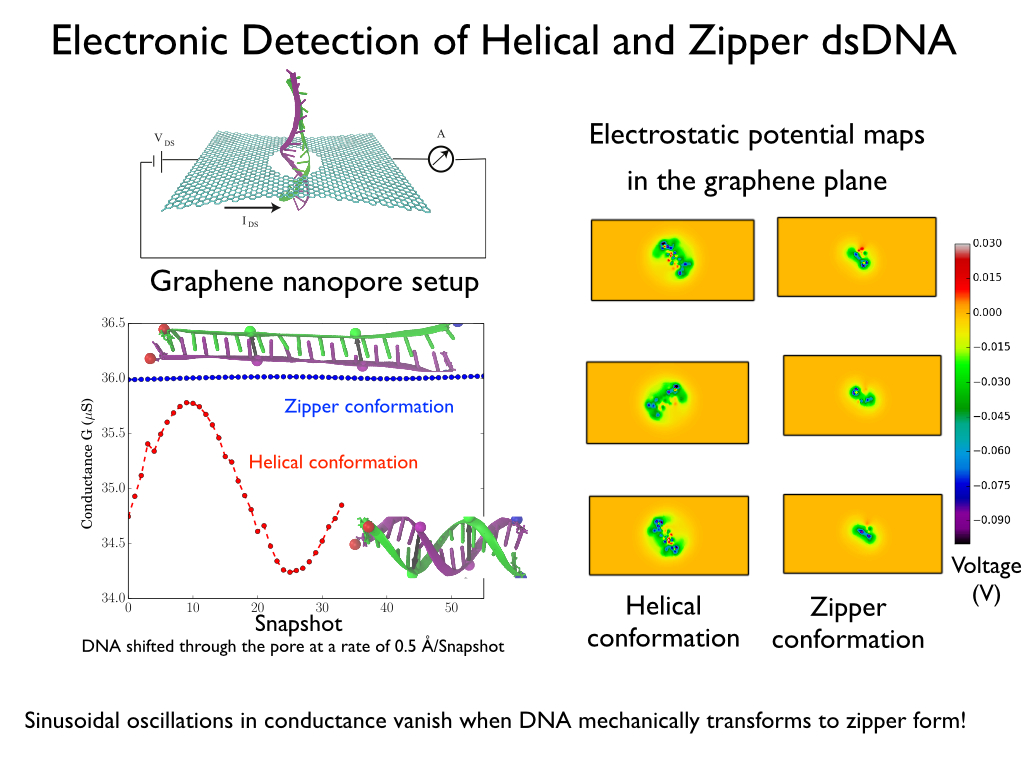
image size:
287.1KB
made with VMD
Threading DNA through a nanometer-size pore, so called
nanopores, drilled into an ultrathin graphene membrane is a promising
approach to build nanobiosensors for sequencing the human genome. Graphene
nanopores can detect translocating DNA by recording concomitant flow of
charged ions through the pore (see December
2011 highlight). As reported in the December
2013 highlight, graphene, which is an electrical conductor, offers a new
way of sensing DNA molecules by monitoring sheet currents along the graphene
membrane. DNA is a highly extensible molecule and upon mechanical manipulation can
change its structure from a canonical helical conformation to a linear
zipper-like conformation. A new study, which
combines classical molecular dynamics simulations using NAMD with quantum mechanical
simulations, suggests that sheet currents, in graphene membranes, can be used
to detect conformation and sequence of
a DNA molecule passing through the nanopore. This new research will guide the
development of graphene-based nanosensors for DNA detection.
More information can be found on our graphene nanopore website.
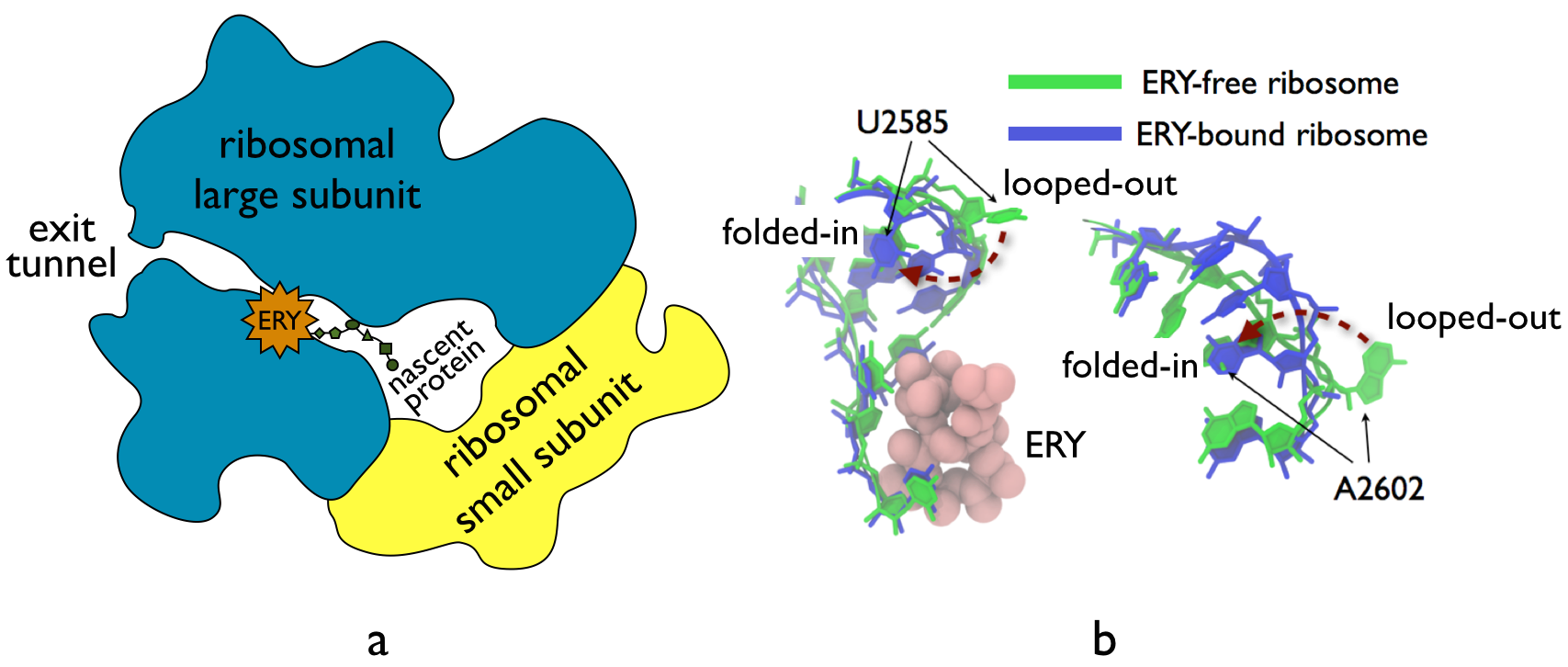
image size:
210.2KB
made with VMD
The ribosome, one of the ubiquitous molecular machines in living cells, is responsible for
the critical task of translating the genetic code into functional proteins (See also
Managing the Protein Assembly Line). The bacterial
ribosome is the target of over 50% antibiotic drugs, for example, the clinically
important macrolide family, including the widely-prescribed erythromycin (ERY) which
is on
the WHO essential medicines list. The antibiotic action of macrolide drugs
has been known for over 50 years, however, the molecular mechanisms underlying the
effects of these drugs are still unknown. It was previously believed that the
antibiotic action by macrolide drugs has to be assisted by the presence of a nascent
protein inside the ribosome. However,
in a
recent study, computational investigations jointly with biochemical
experiments have revealed that the macrolide drugs can take an antibiotic action
by altering the structure of the bacterial ribosome before translation of
nascent protein really begins. Please see more highlights on translational control of
the ribosome: Born to
Control, Shutting Down
the Protein Factory.
Read more on our Ribosome website.
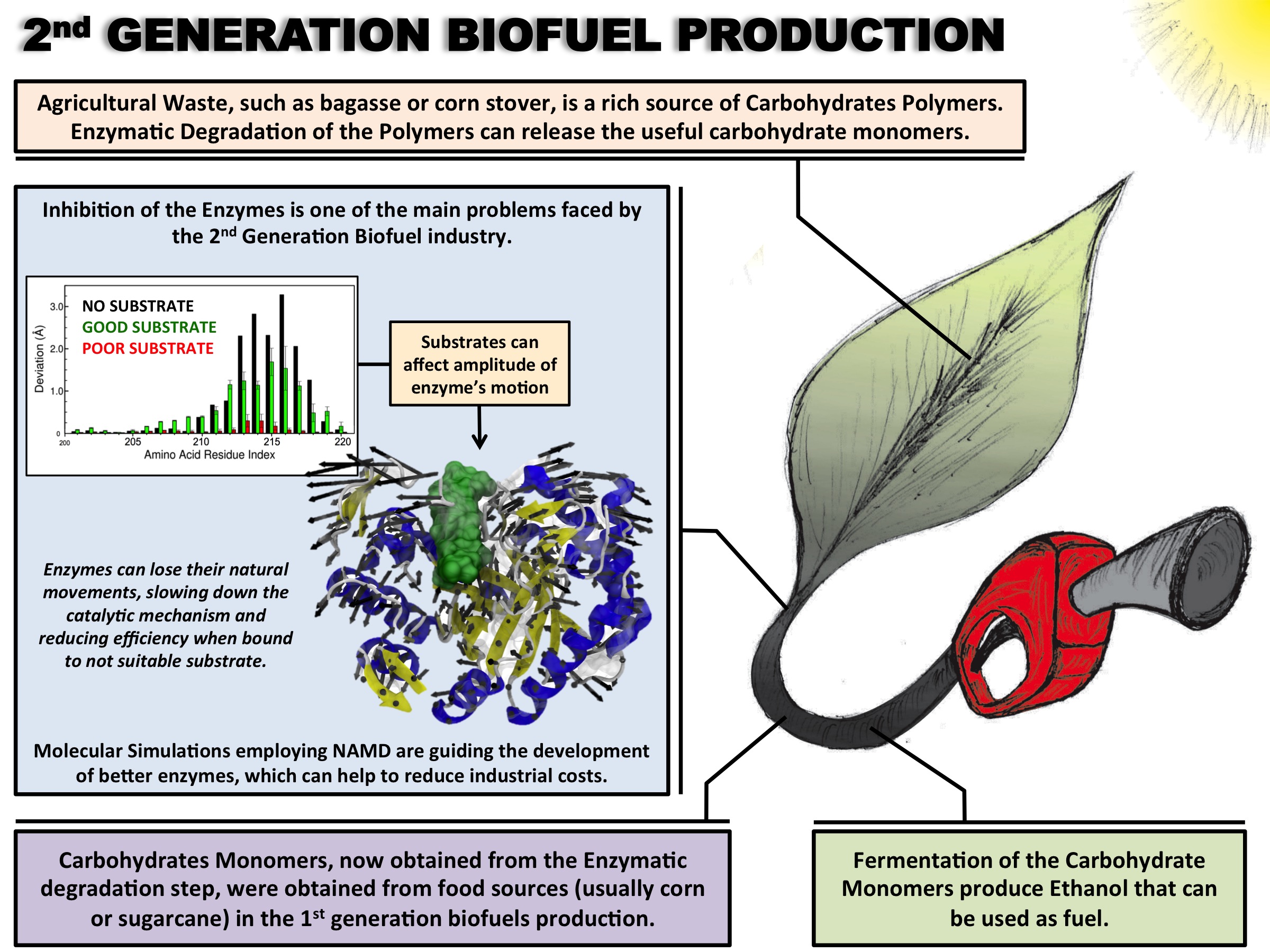
image size: 620.6KB
Biofuels are a well-known alternative to the largely used fossil-derived fuels, however the competition with food production is an ethical dilemma. Fortunately a solution is offered by second-generation biofuels, which can be produced from agricultural waste, or more specifically, from plant cell wall polysaccharides. Using the strategy of microorganisms, several enzymes are employed in the production of this advanced biofuel. However the biofuel industry faces problems such as the loss of efficiency of the enzymes that arises over time due to intermittent high concentration of non-suitable substrates. Simulations can guide biochemical experiments aimed at investigating the mechanism that makes the enzymes vulnerable to such substrates, helping the development of more efficient and, thereby, less costly enzymes. A recent study, based on molecular modeling with NAMD, reported that reduction of efficiency in an important enzyme, know as Man5B, is associated with a loss of the enzyme's flexibility. Molecular dynamis simulations showed that a poor substrate slows down a crucial opening and closing movement of the enzyme's catalytic cleft while a good substrate keeps the movement almost intact. The insight is of crucial importance since it suggests mutations to enzymes presently employed in second-generation biofuel production that need to be replaced less often and, thereby, rendering the production more cost-effective. Read more on our biofuels website.
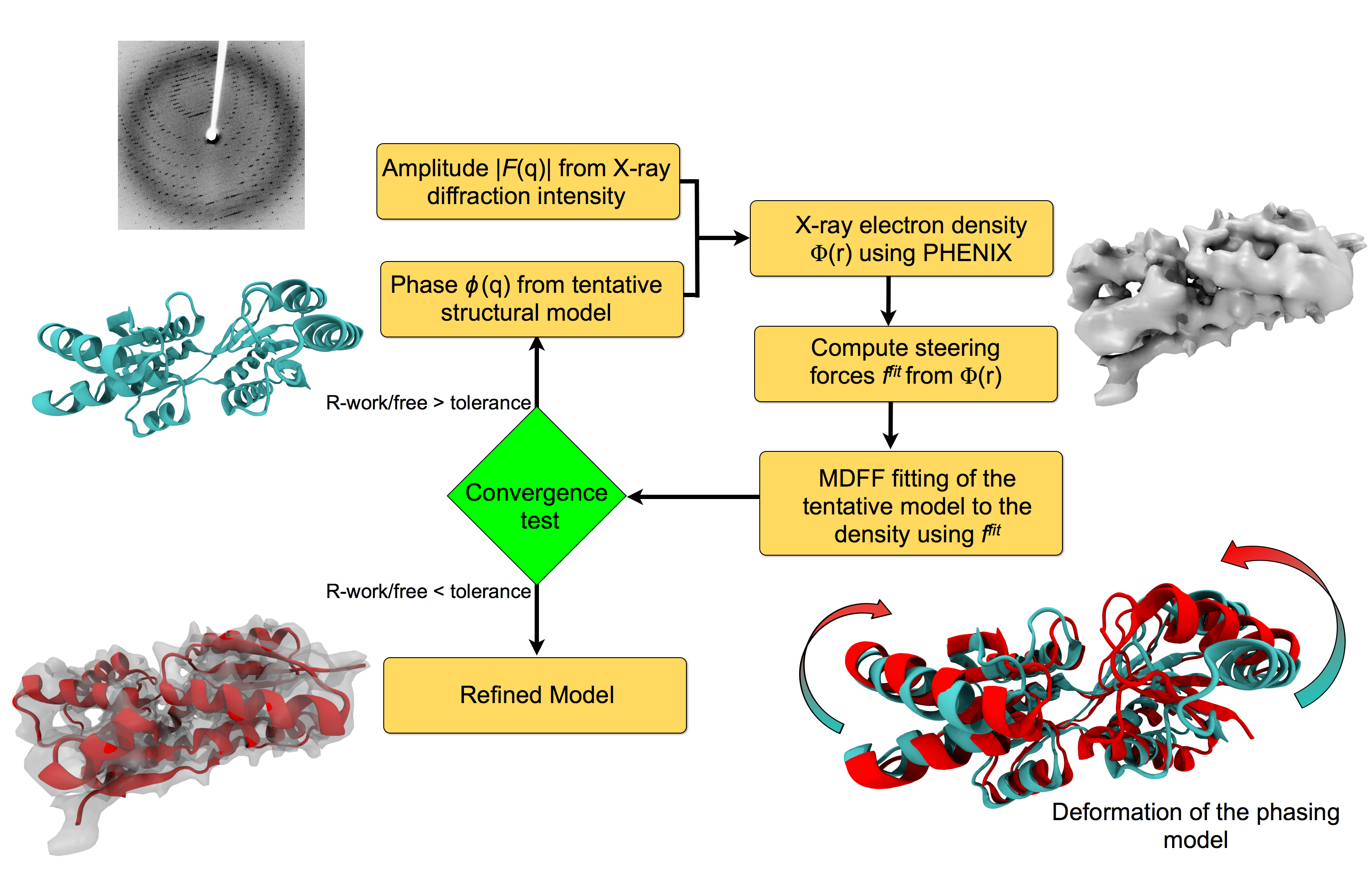
image size:
151.9KB
made with VMD
For many, the word 'X-ray' conjures up the images of white bones on black
backgrounds hanging on the wall of a doctor's office.
However, X-rays have played another important role for the past 100 years
through their use in the determination of chemical structures
at atomic level detail, starting with the first ever structure of
table salt in 1924. Since then, the diffraction properties of X-rays, when
shone on a crystal, have been used to solve increasingly large
and complex structures including those of biological macromolecules found inside living cells.
X-ray crystallography has become the most versatile and dominant technique for
determining atomic structures of biomolecules, but despite its strengths, X-ray crystallography struggles
in the case of large or flexible structures as well as in the case of membrane proteins, either of which diffract only at low resolutions.
Because solving structures from low-resolution
data is a difficult, time-consuming process, such data sets are often discarded.
To face the challenges posed by low-resolution,
new methods, such as xMDFF (Molecular Dynamics Flexible Fitting
for X-ray Crystallography) described
here,
are being developed.
xMDFF extends
the popular MDFF software originally created for determining atomic-resolution structures from
cryo-electron microscopy density maps (see the previous highlights
Seeing Molecular Machines in Action,
Open Sesame,
Placing New Proteins, and
Elusive HIV-1 Capsid).
xMDFF provides a relatively easy solution to the difficult process of refining structures from
low-resolution data. The method has been
successfully applied to experimental data as described in a
recent article
where xMDFF refinement is explained in detail and its use is demonstrated.
Together with electrophysiology experiments, xMDFF was also used to validate
the first all-atom structure of the voltage sensing protein Ci-VSP, as also
recently reported.
More on our
MDFF website.
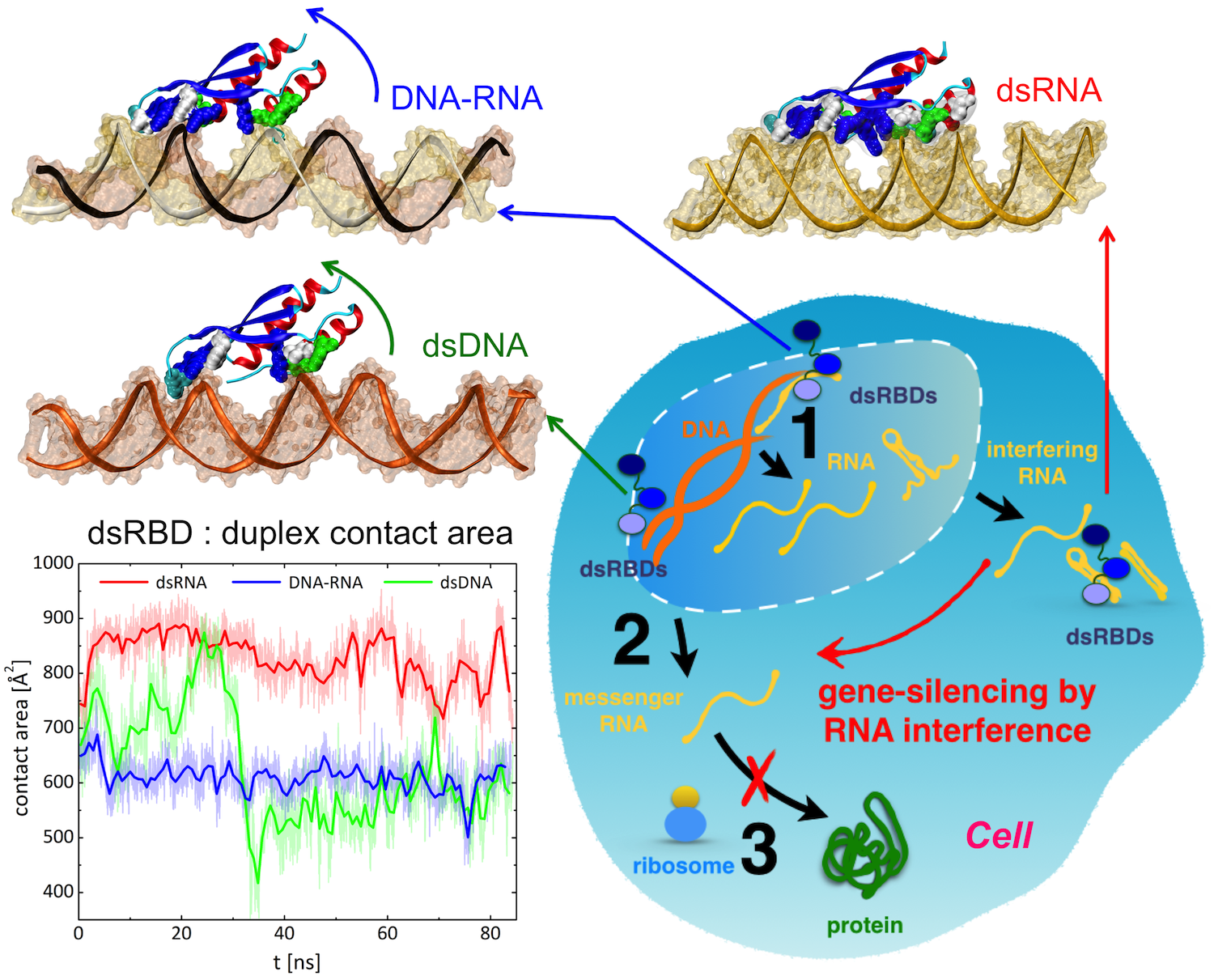
image size:
1.6MB
made with VMD
Only 2% of human DNA involves genes that code for proteins, the machinery of
our cells. The DNA with genes is copied into mRNA read by ribosomes that synthesize then the
respective proteins. However, 80% of the remainder DNA is also copied into
RNA molecules that assume then manifold functions, one being that the RNA
molecules bind to mRNA and silence thereby the respective genes. Frequently,
gene-silencing RNA is activated with the help of proteins called double
stranded RNA binding domains (dsRBDs). These domains bind to parts of
gene-silencing RNA that happens to form double strands, similar but not
identical to the double strands formed by DNA and discovered long ago by Watson
and Crick. In fact, there exist small but characteristic differences between
double stranded RNA, double stranded DNA and hybrid double strands made of RNA
and DNA. In a
recent study, computational biologists performed simulations using the
molecular dynamics program NAMD to determine how dsRBDs
recognize silencing RNA and discriminate between double stranded RNA, DNA, and
hybrid double strands. The simulations revealed that dsRBDs and double stranded
RNA fit together ideally like matching pieces of a puzzle, with mutually
compatible shapes and electrostatic patterns. On the other hand, dsRBDs and
double stranded DNA or hybrid double strands have poor fits due to changed and
insufficiently flexible double strand forms. More
here.
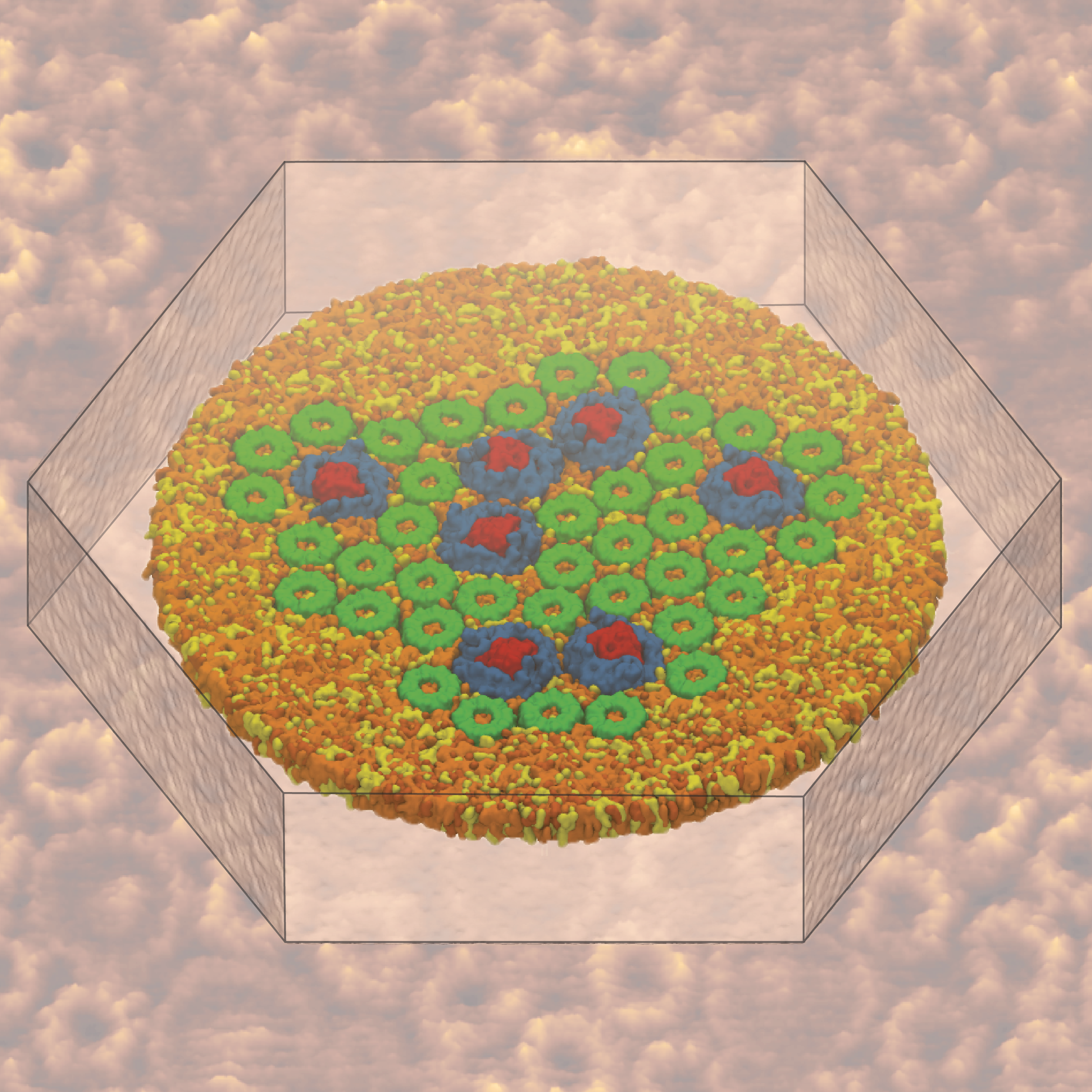
Atomic force microscopy (AFM) gives us a low-resolution glimpse of life at the nanometer scale. Now scientists can bring microscopy images to life by combining the microscopy data with atomic-detail structures to re-create the imaged system on the computer. As recently reported, Center scientists constructed an atomic-resolution model of a photosynthetic membrane based on AFM data showing the locations of the many light-harvesting proteins that inhabit the membrane. After using NAMD on petascale computers, like Blue Waters and Titan, to relax the 20-million atom membrane, Center scientists used the model to study the migration of energy among the light-harvesting complexes, as well as the mobility of quinone molecules in the membrane. The photosynthetic membrane patch was found to have a very high (90%) light-harvesting efficiency; further, it was found that the light-harvesting proteins could be considerably less tightly packed in the membrane with minimal loss of efficiency. Read more on our website.
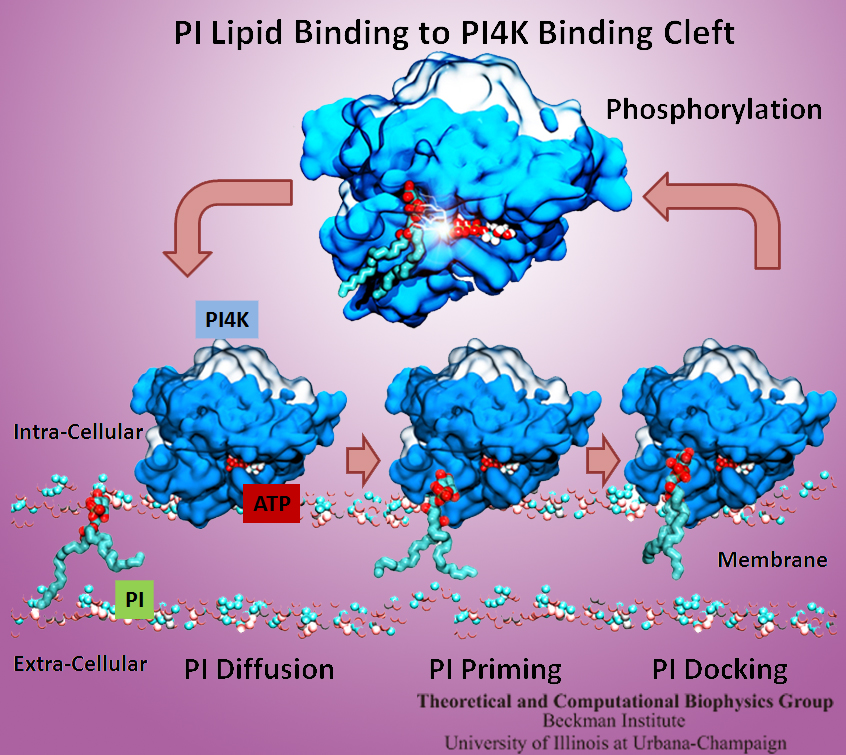
The Golgi apparatus found in so-called eukaryotic cells acts like Amazon.com, namely accepting delivery of newly synthesized proteins, packaging them, and sending them out.
However, in comparison to Amazon.com the Golgi apparatus uses immensely more advanced packaging materials made of a multitude of lipids.
The various lipids form membranes in the shape of vesicles.
Depending on lipid type specific goods are packaged inside the vesicles, specific
locations in the cell receive the packages, content is emptied there and packages are retrieved.
To achieve the series of steps just outlined, lipids as the main actors need to be coordinated.
One way is to recognize lipids forming vesicle membranes and to modify them to be readied for a subsequent step, for example going from release step to retrieval step.
For this purpose eukaryotic cells engage a special class of proteins,
named kinases, that can recognize membrane lipids and phosphorylate them, adding a so-called phosphate group.
As reported recently,
a team of experimental and computational scientists determined the atomic structure of a key member in the kinase family,
phosphatidylinositol 4-kinase (PI4K).
The scientists discovered not only the structure, but also how PI4K captures and phosphorylates a particular type of lipid molecule,
thereby changing a vesicular membrane and turning on the next step in the cellular package delivery system.
The discoveries, made possible through the software
NAMD and VMD,
are expected to have an impact on the design of novel drugs that suppress cancer cell growth. More on our kinase website.
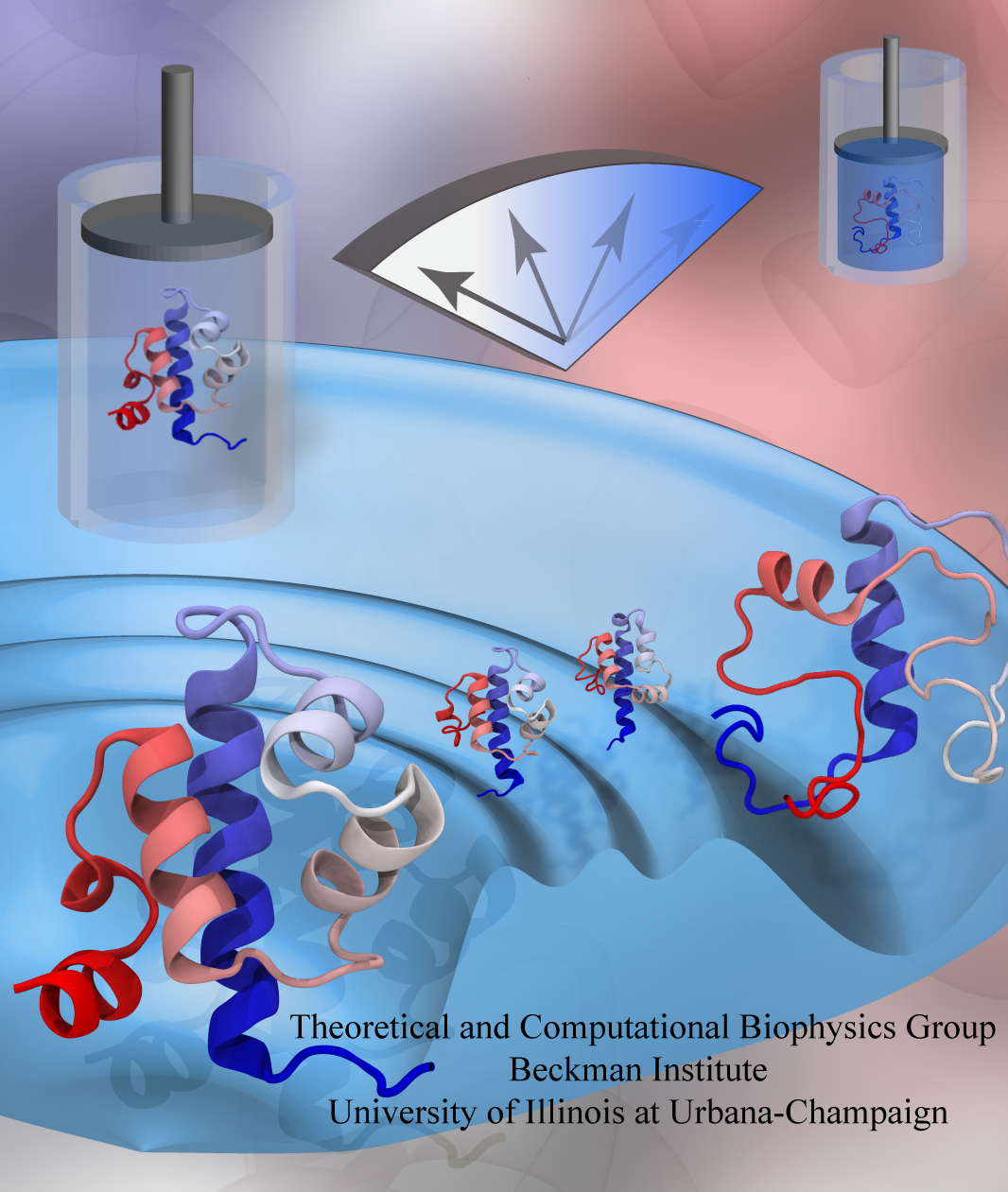
While pressure can help in cooking your favorite meat for dinner,
pressure is also helping scientists to study how proteins,
a key ingredient in any meal, loose and regain their proper shape.
Proteins are key building blocks for any life form on earth,
making the many machines that drive living cells.
For any protein to do its job correctly,
it has to first settle into the proper shape, the so-called native state.
The process, referred to as protein folding, is still a mystery
(see
July 2012,
Nov 2009 and
May 2008 highlights).
A general consensus is that the mystery can be solved only through a combination of experimental observation and computer simulation.
In two recent reports
(1 and
2),
a team of experimental and computational scientists have used high pressure to force a protein to loose its proper shape,
similar to what happens in a pressure cooker.
After the high pressure is released in the experiment, the protein regains its proper shape,
apparently by following two folding pathways,
one fast and one slow.
Using the molecular dynamics simulation program
NAMD,
as well as a special purpose supercomputer,
Anton,
the researchers were able to identify these two pathways and to follow every single folding step of the protein at an unprecedented precision.
The studies greatly improve scientific understanding at the molecular-level of how proteins respond to pressure changes and,
while not giving delicious recipes for pressure cooking in the kitchen,
they are likely to suggest how to use pressure to dissolve toxic proteins that arise in disease,
such as in Alzheimer's disease.
More on our protein folding website.
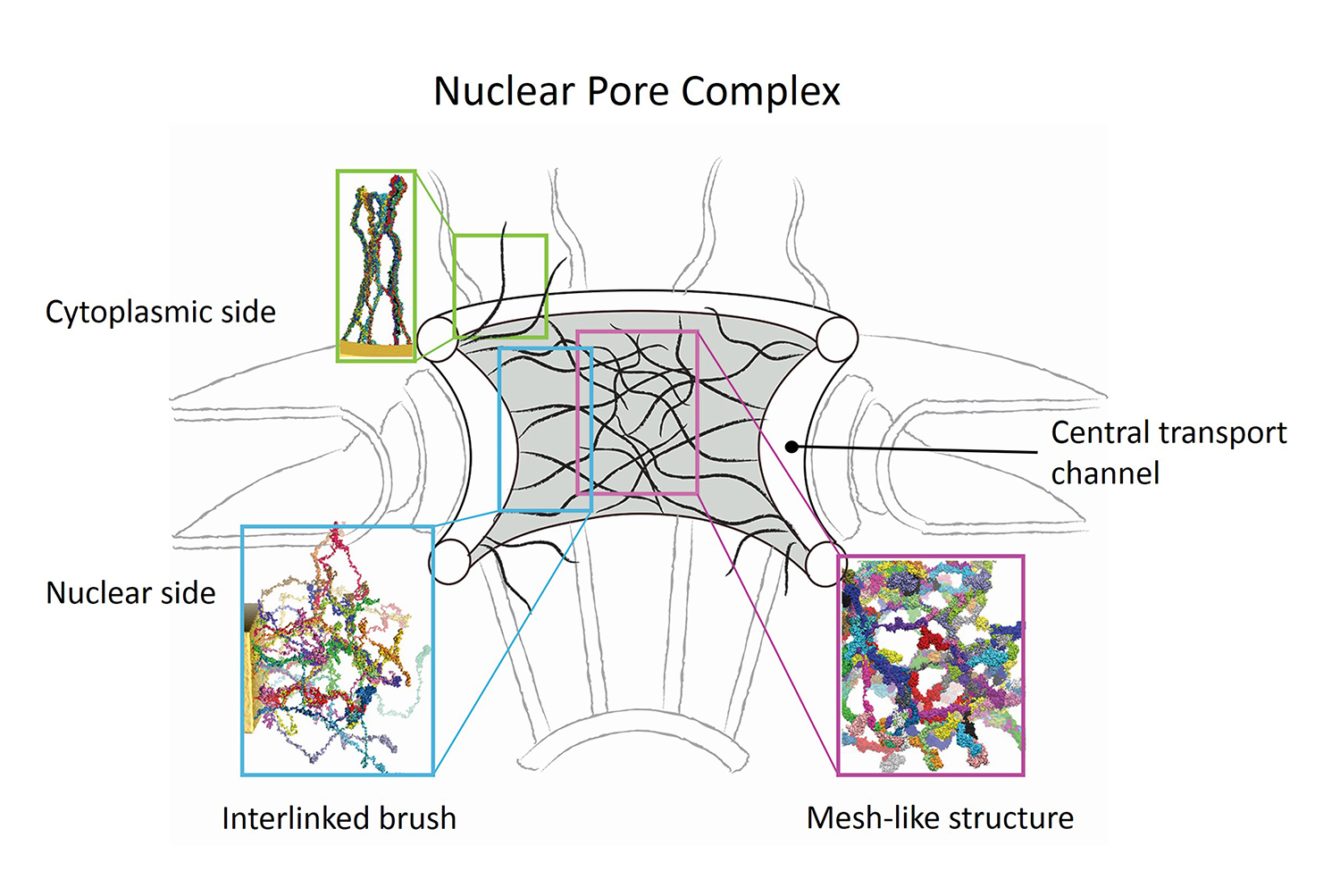
image size:
579.0KB
made with VMD
Nanoengineers building nanodevices achieve technological solutions at scales of 100 nanometers or 0.0001 mm.
Nanoengineering is a brand new human technology, just a few decades old.
In living cells, nanoengineering solutions are actually a few
billion years old and therefore much more intricate.
An impressive example is the nuclear pore,
hundreds to thousands of which dot the nuclear membrane
that separates in eukaryotic cells the genome and its molecular control factors from the cytoplasm of the cell.
Only since very recently could cell biologists begin to resolve the molecular architecture of the nuclear pore.
Given the pore's many-fold functions, like letting small molecules pass easily,
but larger ones only as cargoes of special proteins,
the transport factors, or adapting the pore size when large cargoes need to pass,
the architecture of the nuclear pore is complex, involving an assembly of hundreds of proteins.
The interior of the pore is filled with 600 amino acid-long "finger" proteins tethered at the periphery.
The finger proteins are largely disordered such that experimental methods
lack resolving power and computational modeling is needed to
figure out their dynamic arrangement and traffic control function,
but such modeling was largely unfeasible; only a small fraction of the nuclear pore
volume could be covered computationally.
The advent of petascale computing increased the size-scale of biomolecular
simulations hundred-fold and a recent report
employing
the programs NAMD and VMD took advantage of the new generation of computers,
simulating the dynamic, disordered arrangement of nuclear pore proteins.
The simulations, still at an early stage, suggest a detailed,
atomic level picture of the nuclear pore interior together
with an explanation of molecular traffic control. More on our nuclear pore website.
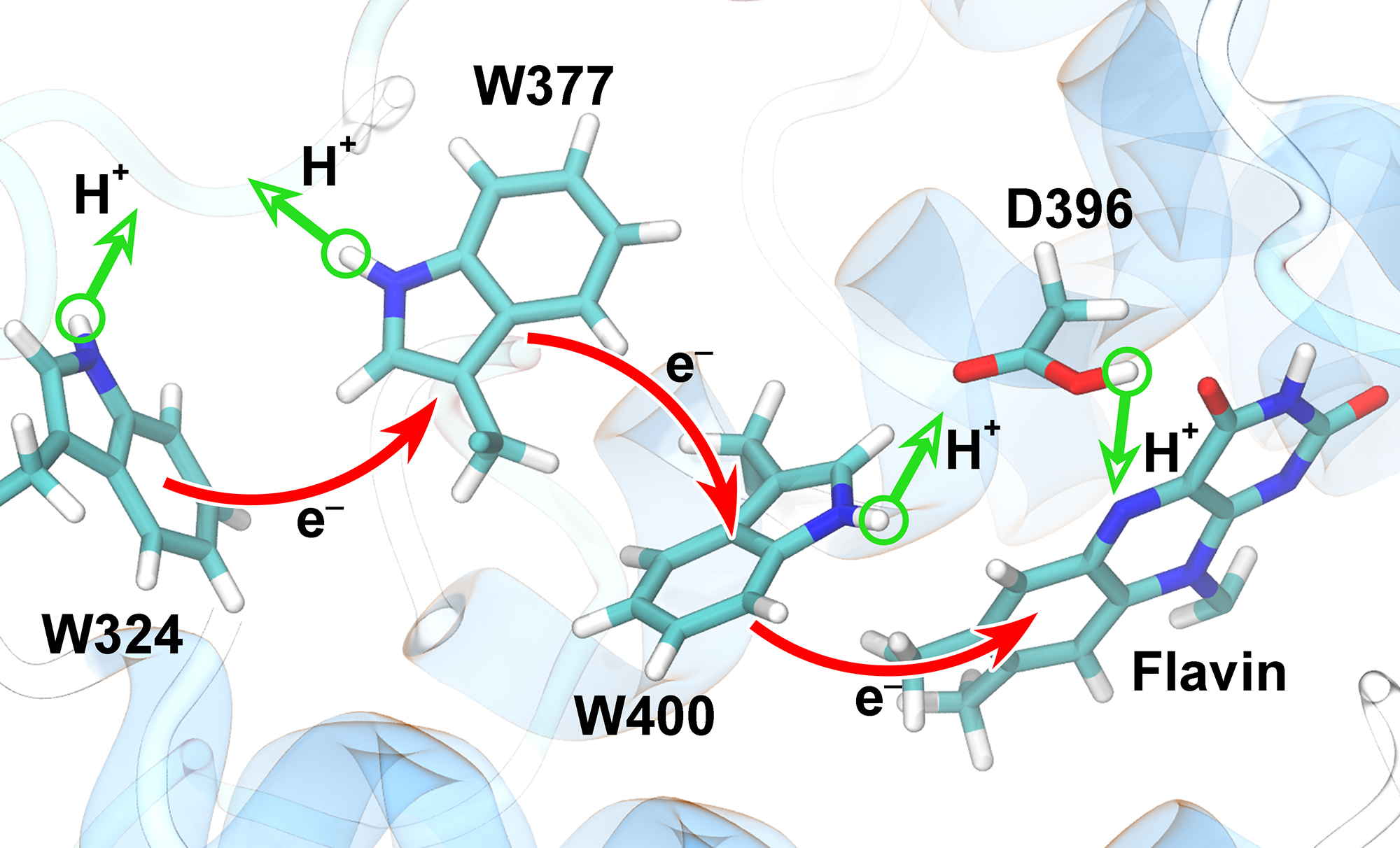
image size:
818.1KB
made with VMD
Animals and plants possess internal clocks that attune them to the daily or solar as well as to the tidal or lunar rhythm on Earth. A key blue-light receptor serving for this purpose is a protein called cryptochrome. Another apparent role of cryptochrome is that of a sensor for the geomagnetic field. In this role cryptochrome assists migratory birds in long-range navigation
(see February 2012,
July 2010,
July 2009, and
April 2007 highlights).
The magnetoreceptor function of cryptochrome arises from light-induced electron transfer between a flavin cofactor and a tryptophan residue.
A recent study shows now that subsequent further electron transfers yield a pair of so-called entangled, freely to precess, electron spins spaced sufficiently far apart to establish cryptochrome's sensitivity to the geomagnetic field. The study reveals how, through a combination of classical motion of the protein and quantum dynamics of some of its electronic degrees of freedom the sequential electron transfer comes about and leads to the two widely separated electron spins. These spins act much like recently invented quantum computers, except that their role in migratory birds is to act as a light-driven magnetic compass.
More on our cryptochrome webpage.
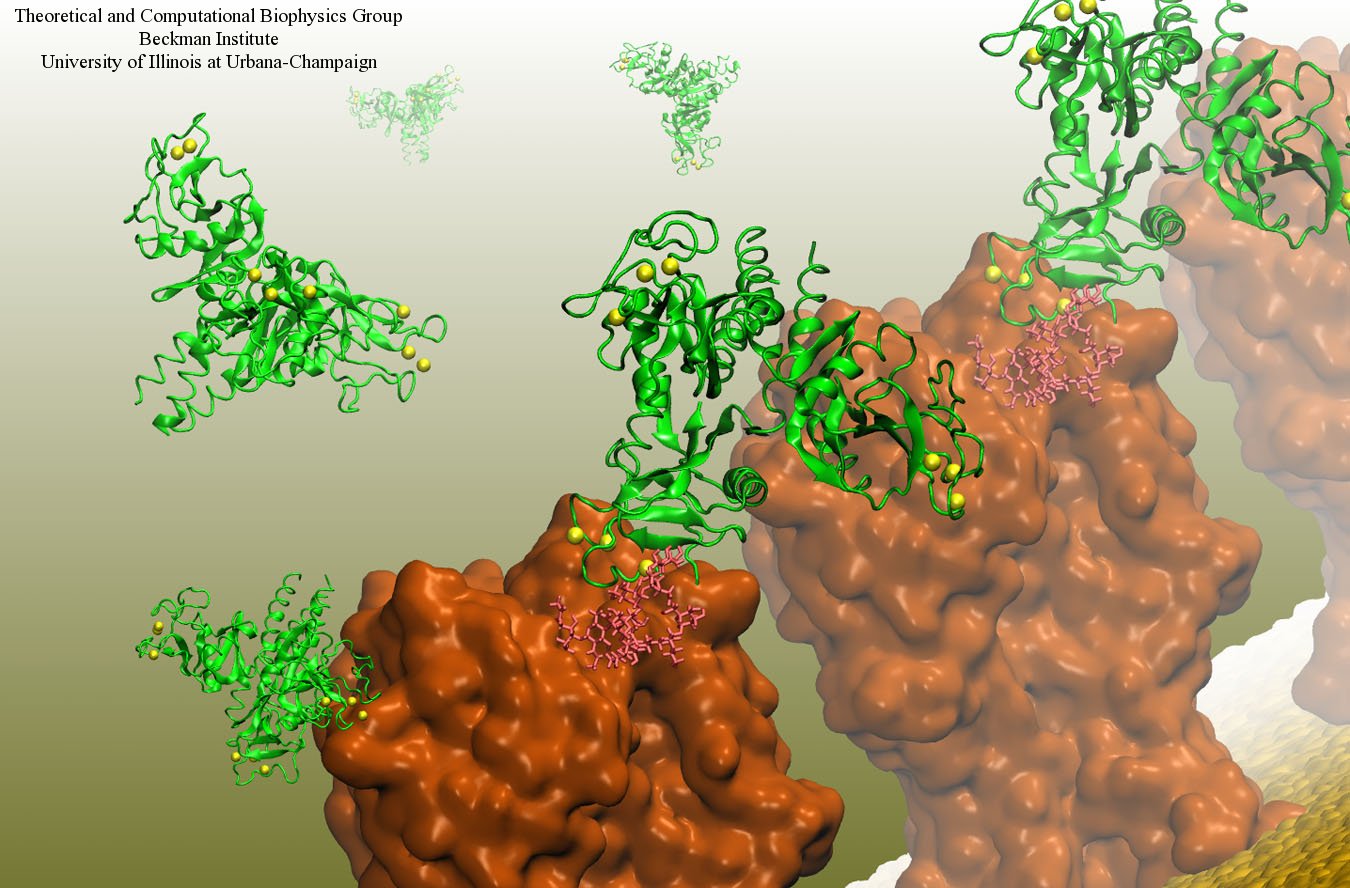
image size:
199.7KB
made with VMD
Our body uses several defense mechanisms against seasonal flu, the common affliction caused by influenza viruses. By taking a yearly flu shot, our body's defense based on antibodies is trained and envoked. A defense system not based on antibodies acts at the very front line of influenza virus attack, namely the lungs.
For this protection the body uses so-called lung surfactant proteins that coat the inner lining of the lungs to keep a wet film on the lung surface needed for oxygen-carbon dioxide exchange. The lung surfactant proteins also serve as police against influenza viruses. For this purpose the lung surfactant protein D (SP-D) recognizes a protein component of the virus surface, namely hemagglutinin, and handcuffs the sugar molecules bound to hemagglutinin. A previous experimental-simulation study (see October 2012 highlight) found that SP-D of pigs exhibits a stronger inhibitory activity against influenza A virus in this regard than does human SP-D. In a recent study, researchers have now boosted the protective ability of human SP-D by introducing mutations. Molecular dynamics simulations using NAMD suggest that the mutated human SP-D employs a different and stronger blocking mechanism on the active site of influenza A virus than native SP-D does. Combined with experimental results, the simulations suggest a mechanism through which SP-D acts, namely, by handcuffing viruses together and, thereby, preventing viral entry into cells. The findings from this research might lead to a new protection against seasonal flu, namely a nasal spray containing mutated lung surfactant proteins that strengthen a person's armada of defense proteins on the lung surface. More on our lung surfactant protein website.


