NAMD, recipient of a 2002 Gordon Bell Award, a 2012 Sidney Fernbach Award, and a 2020 Gordon Bell Prize, is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. Based on Charm++ parallel objects, NAMD scales to hundreds of cores for typical simulations and beyond 500,000 cores for the largest simulations. NAMD uses the popular molecular graphics program VMD for simulation setup and trajectory analysis, but is also file-compatible with AMBER, CHARMM, and X-PLOR. NAMD is distributed free of charge with source code. You can build NAMD yourself or download binaries for a wide variety of platforms. Our tutorials show you how to use NAMD and VMD for biomolecular modeling.
Breaking News
|
NAMD 3.0.3 Release -
Point release 3.0.3 fixes an incorrect wrapping bug in spinAngle in the Colvars in NAMD 3.0, 3.0.1 and 3.0.2. See announce.txt for details regarding correctness issues in: eABF and any other methods relying on extended Langrangian dynamics. This point release also fixes crashes in the GPU-offload GBIS simulations. We encourage all NAMD users to upgrade to version 3.0.3. |
| NAMD 3.0 New Features - webpage is posted |
| NAMD GPU-resident benchmarks - results and data sets with GPU-optimized configuration posted |
| NAMD 2.15 ALPHA Release providing GPU-offload support for Intel GPU Max Series. This source code release available on the download page includes SYCL code that can be built using the Intel oneAPI toolkits. Following the download link reveals a page with detailed build instructions. |
Spotlight: May the Force Field Be With You (Sept 2013)
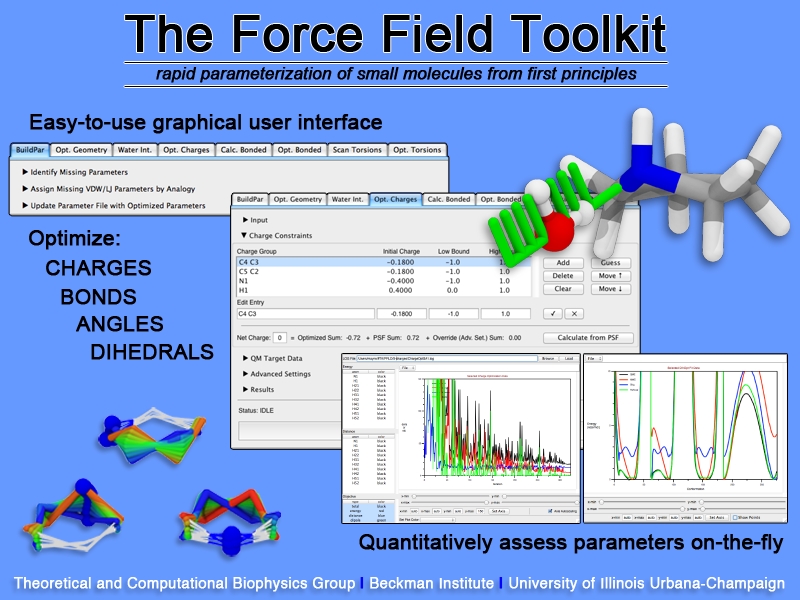
image size:
307.6KB
Structural biologists are increasingly turning to simulation methods to investigate the connections between molecular structure and biological function. Classical molecular dynamics (MD) simulations, such as those performed by the simulation software NAMD, rely on potential energy functions requiring parameters to describe atomic interactions within the molecular system. While these parameters are available for the most commonly simulated biopolymers (e.g., proteins, nucleic acids, carbohydrates), many small molecules and other chemical species lack adequate descriptions. The complexity of developing these parameters severly restricts the application of MD technologies across many fields, including most notably drug discovery. Recently, researchers have developed software, the Force Field Toolkit (ffTK), that greatly reduces these limitations by facilitating the development of parameters directly from first principles. ffTK, distributed as a plugin for the molecular modeling softare VMD, addresses both theoretical and practical aspects of parameterization by automating tedious and error-prone steps, performing multidimensional optimizations, and providing quantitative assessment of parameter performance--all from within an easy-to-use graphical user interface. Additional information on ffTK, including documentation and screencast tutorials, can be found here.
Overview
Why NAMD? (in pictures)
How to Cite NAMD
Features and Capabilities
Performance Benchmarks
Publications and
Citations
Credits and Development Team
Availability
Read the License
Download NAMD Binaries
(also VMD)
Build from Source Code
- Git access now available
Run at NCSA, SDSC, NICS, or Texas
Training
NAMD Developer Workshop in Urbana (August 19-20, 2019)
PRACE School on HPC for Life Sciences (June 10-13, 2019)
"Hands-On" Workshop in Pittsburgh (May 13-17, 2019)
Charm++ Workshop in Urbana (May 1-2, 2019)
Enhanced Sampling and Free-Energy Workshop (Sept 10-14, 2018)
NAMD Developer Workshop in Urbana (June 11-12, 2018)
"Hands-On" Workshop in Pittsburgh (May 21-25, 2018)
"Hands-On" QM/MM Simulation Workshop (April 5-7, 2018)
Older "Hands-On" Workshops
Support
Mailing List Issues for Yahoo.com Addresses
Announcements
NAMD 3.0.3 Release (Jul 2026)
NAMD 3.0.2 Release (Aug 2025)
NAMD 3.0.1 Release (Oct 2024)
NAMD 3.0 Release (Jun 2024)
NAMD 3.0 New Features (Feb 2024)
NAMD 2.14 Bug Fixes (Apr 2022)
NAMD 2.14 Release (Aug 2020)
NAMD 2.14 New Features
One-click NAMD/VMD in the cloud
QM/MM Interface to MOPAC and ORCA
QwikMD GUI Released in VMD 1.9.3
Previous Announcements
Documentation
Related Codes, Scripts, and Examples
NAMD Wiki (Recent Changes)
Older Documentation
News
Sparing healthy microbes while using a novel antibiotic
AMBER force field use in NAMD for large scale simulation
NAMD GPU-resident benchmarks available
NAMD and VMD share in COVID-19 Gordon Bell Special Prize
NAMD reference paper published online
Coronavirus Simulations by U. Delaware Team
Coronavirus Simulations on Frontera Supercomputer
Breakthrough Flu Simulations
Oak Ridge Exascale Readiness Program
Prepping for Next-Generation Cray at NERSC
Supercomputing HIV-1 Replication
How GPUs help in the fight against staph infections
Computational Microscope Gets Subatomic Resolution
Opening New Frontiers in the Battle Against HIV/AIDS
HIV Capsid Interacting with Environment
Assembling Life's Molecular Motor
Older News Items


