Over 3,000 citations of NAMD reference paper
July 5, 2013
The NAMD developers thank our users for 3,000 citations of our 2005 reference paper:
James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kale, and Klaus Schulten. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26:1781-1802, 2005.
NAMD is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD scales to hundreds of processors on high-end parallel platforms, as well as tens of processors on low-cost commodity clusters, and also runs on individual desktop and laptop computers. NAMD works with AMBER and CHARMM potential functions, parameters, and file formats. This article, directed to novices as well as experts, first introduces concepts and methods used in the NAMD program, describing the classical molecular dynamics force field, equations of motion, and integration methods along with the efficient electrostatics evaluation algorithms employed and temperature and pressure controls used. Features for steering the simulation across barriers and for calculating both alchemical and conformational free energy differences are presented. The motivations for and a roadmap to the internal design of NAMD, implemented in C++ and based on Charm++ parallel objects, are outlined. The factors affecting the serial and parallel performance of a simulation are discussed. Finally, typical NAMD use is illustrated with representative applications to a small, a medium, and a large biomolecular system, highlighting particular features of NAMD, for example, the Tcl scripting language. The article also provides a list of the key features of NAMD and discusses the benefits of combining NAMD with the molecular graphics/sequence analysis software VMD and the grid computing/collaboratory software BioCoRE. NAMD is distributed free of charge with source code at www.ks.uiuc.edu.
This paper is available from our website or directly from the journal.
The NAMD License Agreement requires that this paper be cited by any published work which utilizes NAMD. Proper citation is essential to continued NIH funding for NAMD development, as it is a primary way in which we demonstrate the value of our software to the scientific community. As of May 2013, over 3900 publications have cited either the 1999 or 2005 reference papers.
Below are a few of the many biomedically relevant discoveries made by internal and external NAMD users since our March 2010 milestone of 1,000 citations.
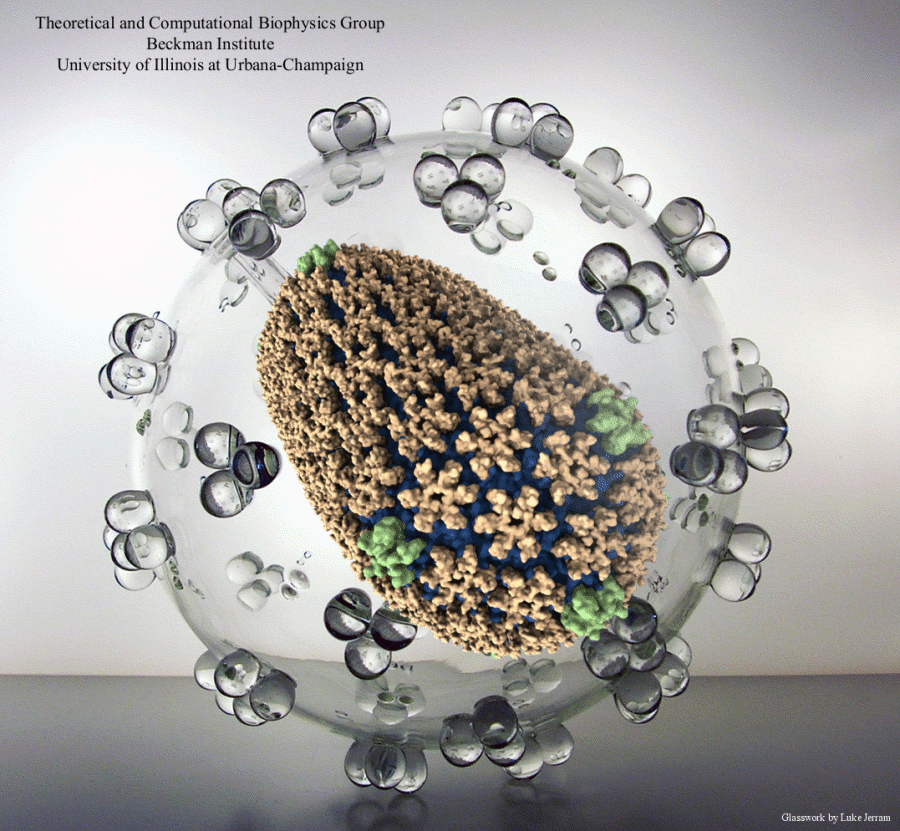
Human immunodeficiency virus type 1 (HIV-1) is the major cause of AIDS, for which treatments need to be developed continuously as the virus becomes quickly resistant to new drugs. When the virus infects a human cell it releases into the cell its capsid, a closed, stable container protecting the viral genetic material. However, interaction with the cell triggers at some point an instability of the capsid, leading to a well timed release of the genetic material that merges then with the cell's genes and begins to control the cell. The dual role of the capsid, to be functionally both stable and unstable, makes it in principle an ideal target for antiviral drugs and, in fact, treatments of other viral infections successfully target the respective capsids. The size of the HIV-1 capsid (about 1,300 proteins), and its irregular shape had prevented so far the resolution of a full capsid atomic-level structure. However, in a tour de force effort, groups of experimental and computational scientists have now resolved the capsid's chemical structure (deposited to the protein data bank under the accession codes 3J3Q and 3J3Y). As reported recently (see also journal cover), the researchers combined NMR structure analysis, electron microscopy and data-guided molecular dynamics simulations utilizing VMD to prepare and analyze simulations performed using NAMD on one of the most powerful computers worldwide, Blue Waters, to obtain and characterize the HIV-1 capsid. The discovery can guide now the design of novel drugs for enhanced antiviral therapy. More information is available on our virus website, in video, and in a press release.

Rabbit hemorrhagic disease is extremely contagious and associated with liver necrosis, hemorrhaging, and high mortality in adult rabbits. First described in China in 1984, within a few years, rabbit hemorrhagic disease spread to large parts of the world and today threatens the rabbit industry and related ecology. The disease is caused by a virus, aptly named rabbit hemorrhagic disease virus. As reported recently, a group of experimental and computational researchers combining crystallography, electron microscopy and data-guided molecular dynamics simulations utilizing NAMD determined an atomic model of the capsid, namely the protein shell that surrounds the genetic material of the virus. The capsid simulations involved 10 million atoms and have become feasible only through Blue Waters, a brand new petascale supercomputer. The atomic model, analyzed by means of VMD, recently adapted to studies of very large structures, resolves the structural framework that furnishes both mechanical protection to the viral genes as well as a quick release mechanism after a virus enters a host cell. Researchers can use the detailed knowledge of the capsid structure to develop vaccines against rabbit hemorrhagic disease. More information is available on our virus website and in a news story.
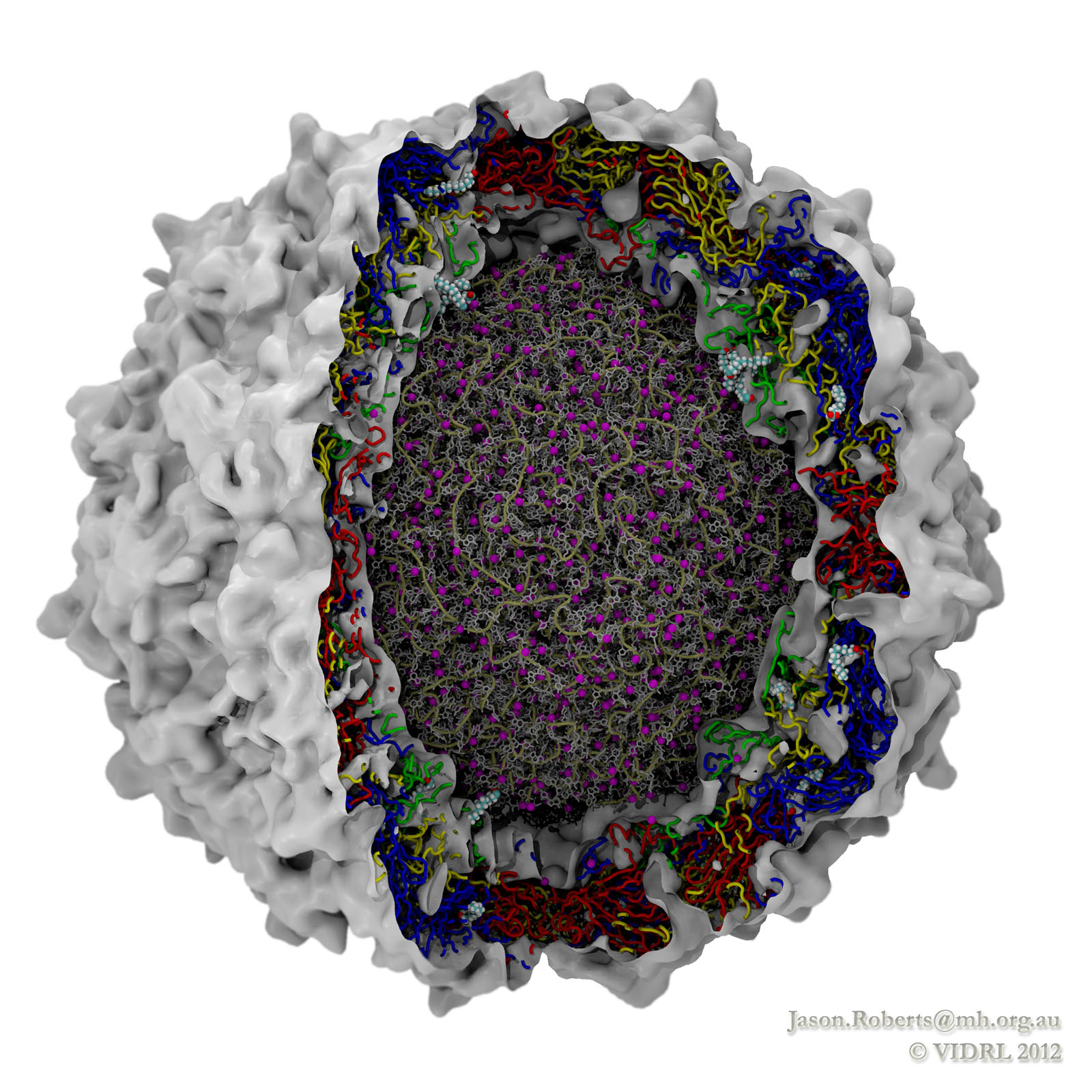
While a global campaign to eradicate polio has resulted in only 212 cases reported worldwide in 2012, there is a continuing risk of vaccine-resistant strains emerging. In addition, as one of the most studied viruses of all time, polio makes an excellent model for understanding similar diseases. Researchers at the Victorian Infectious Diseases Reference Laboratory have reconstructed and simulated a complete polio virus, including both the genomic payload RNA core and its surrounding protein capsid. The polio virus model is being used as a basis for understanding antiviral drugs, virus infection, and to learn how to model related viruses such as Enterovirus 71. The model was constructed in VMD and simulations of 3.7 million atoms were performed using NAMD on the BlueGene/Q supercomputer at the Victorian Life Sciences Computation Initiative. Rhombicdodecahedral periodic boundary conditions were used to reduce the volume of water needed to solvate the virus, and the symmetry of the virus capsid was exploited to study an antiviral drug binding at 60 identical locations simultaneously, avoiding artifacts that could have arisen from a truncated structure. See the researchers' poliovirus simulation website for a movie and additional information.

image size:
77.5KB
made with VMD
Did you get your flu shot this year? Influenza is a leading cause of preventable death in the industrialized world, representing hundreds of billions of dollars in healthcare expenditures and loss of economic production. While the yearly influenza vaccination is nearly 90% effective at limiting infections in populations less than 65 years of age, there is insufficient evidence regarding the effectiveness of the flu shot for the elderly population, whose immune systems may not mount an adequate antibody response to vaccination. Beyond vaccination, front-line therapies such as the neuraminidase inhibitors Tamiflu and Relenza have proven to be of limited effectiveness due to the evolution of drug-resistant influenza mutants. Therefore, a need exists for the development of new therapies to circumvent these resistance mechanisms. Computational biologists employing NAMD and VMD used molecular simulations to uncover the key role that water plays in mediating how well antiviral drugs can bind to proteins of the influenza virus. This investigation, reported recently, reveals that amino-acid mutations responsible for drug resistance act by reshaping the local electric field and also by permitting infiltration of water within the otherwise hydrophobic drug binding pocket. These mutations thus induce drug resistance in much the same way as inverting the polarity of a magnet can repel rather than attract. These findings are expected to help guide the design of novel drugs with increased antiviral efficacy. Additional details about this study can be found here.
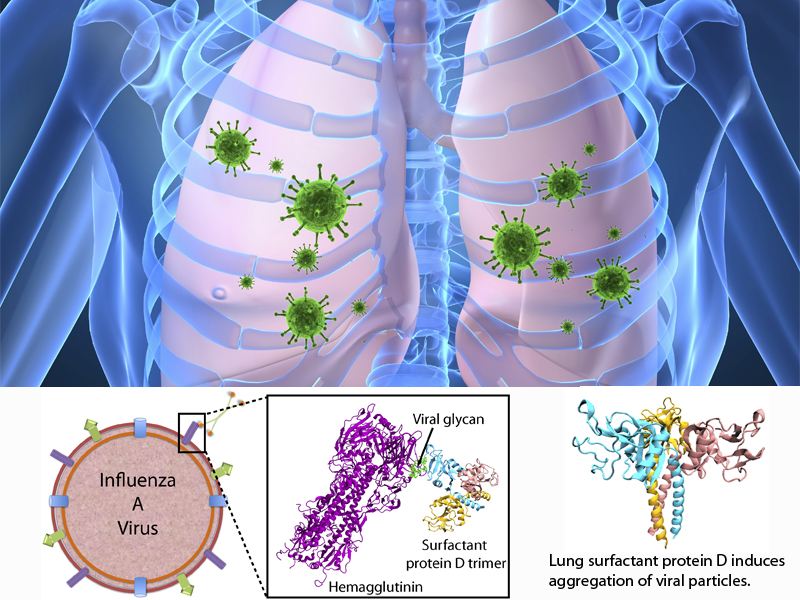
image size:
76.8KB
made with VMD
The fall flu season is coming. It is time to get your flu shot! Many people may still remember the influenza A H1N1 flu ("swine flu") pandemic of 2009, which caused 280,000 deaths worldwide. The best way to prevent the flu is to get vaccinated with a flu shot or use the flu nasal spray vaccine. However, rapid evolution of the flu virus constantly requires new vaccines. Fortunately, the immune system has several defensive mechanisms in the lung to clear inhaled pathogens. One of these mechanisms involves surfactant proteins which induce aggregation of viral particles and, thereby, prevent infection, serving as a front-line host defense. Recently, researchers found that surfactant protein D (SP-D) from pigs exhibits particularly strong inhibitory activity, more so that human SP-D. This discovery leads researchers to investigate SP-D structure-related antiviral activity. In a recent experimental-simulation study, crystallographic analysis of pig and human SP-D showed that a loop involved in viral binding on pig SP-D is longer than the respective loop on human SP-D; molecular dynamics simulation revealed that the longer loop of pig SP-D has higher flexibility than that of human SP-D, suggesting that the flexible loop region could facilitate strong binding of SP-D to virus. Based on this finding, one can develop new nasal spray anti-flu protection through other structural modification of human lung surfactant proteins. More on our lung surfactant protein website.
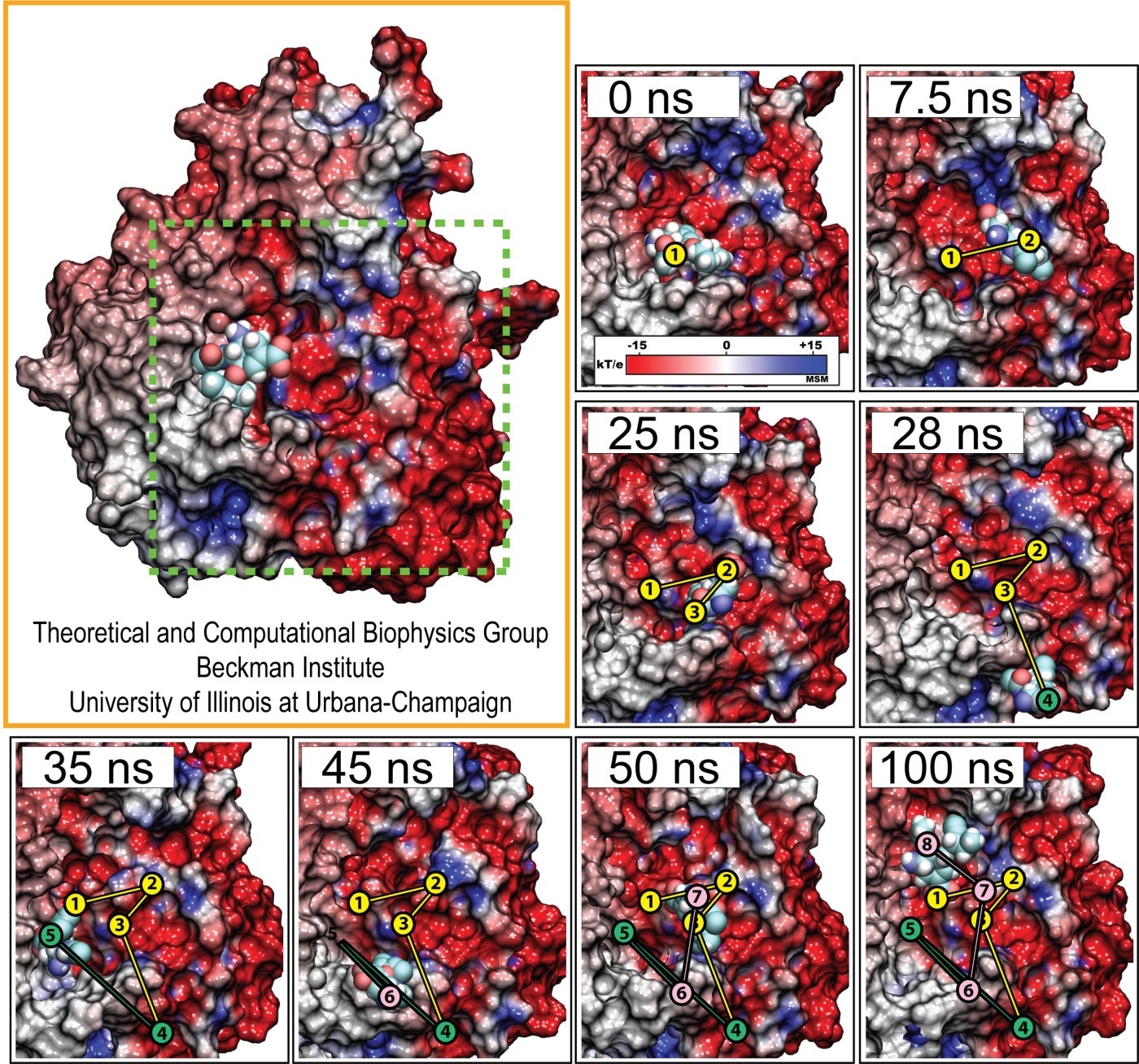
Fever, chills, sore throat, coughing, aches, and pains? Ah ..... you have the flu! As a measure of prevention, vaccines against seasonal influenza are distributed and administered each fall. Last year though, the outbreak of the H1N1pdm "swine" influenza virus, caught health workers by surprise as this virus not only infected individuals during the spring and summer months, but also seemed to be particularly virulent towards otherwise healthy young people. Even more alarming was increasing evidence that H1N1pdm had acquired resistance to the frontline antiflu drug, Tamiflu. In response to this, computational biologists at the University of Illinois and the University of Utah teamed up to uncover the basis for influenza drug resistance through quantum chemistry, and molecular dynamics simulations with NAMD. The results of this study have recently been reported, and uncovered a two stage binding pathway for Tamiflu in H1N1pdm "swine" and H5N1 "avian" flu proteins, as well as a possible mechanism through which genetic mutations can induce drug resistance in one of the stages. Subsequent efforts at drug design against influenza can take advantage of this discovery. This discovery was made possible through use of so-called GPU computing (see Oct 2007 highlight "Graphics Processors Speed Up Simulations"). More information can be found here.


