Over 4,000 citations of NAMD reference paper
September 5, 2014
The NAMD developers thank our users for 4,000 citations of our 2005 reference paper:
James C. Phillips, Rosemary Braun, Wei Wang, James Gumbart, Emad Tajkhorshid, Elizabeth Villa, Christophe Chipot, Robert D. Skeel, Laxmikant Kale, and Klaus Schulten. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26:1781-1802, 2005.
NAMD is a parallel molecular dynamics code designed for high-performance simulation of large biomolecular systems. NAMD scales to hundreds of processors on high-end parallel platforms, as well as tens of processors on low-cost commodity clusters, and also runs on individual desktop and laptop computers. NAMD works with AMBER and CHARMM potential functions, parameters, and file formats. This article, directed to novices as well as experts, first introduces concepts and methods used in the NAMD program, describing the classical molecular dynamics force field, equations of motion, and integration methods along with the efficient electrostatics evaluation algorithms employed and temperature and pressure controls used. Features for steering the simulation across barriers and for calculating both alchemical and conformational free energy differences are presented. The motivations for and a roadmap to the internal design of NAMD, implemented in C++ and based on Charm++ parallel objects, are outlined. The factors affecting the serial and parallel performance of a simulation are discussed. Finally, typical NAMD use is illustrated with representative applications to a small, a medium, and a large biomolecular system, highlighting particular features of NAMD, for example, the Tcl scripting language. The article also provides a list of the key features of NAMD and discusses the benefits of combining NAMD with the molecular graphics/sequence analysis software VMD and the grid computing/collaboratory software BioCoRE. NAMD is distributed free of charge with source code at www.ks.uiuc.edu.
This paper is available from our website or directly from the journal.
The NAMD License Agreement requires that this paper be cited by any published work which utilizes NAMD. Proper citation is essential to continued NIH funding for NAMD development, as it is a primary way in which we demonstrate the value of our software to the scientific community. As of April 2014, over 4800 publications have cited either the 1999 or 2005 reference papers.
Below are a few of the many biomedically, environmentally, and methodologically relevant discoveries made by NAMD users since our July 2013 milestone of 3,000 citations.
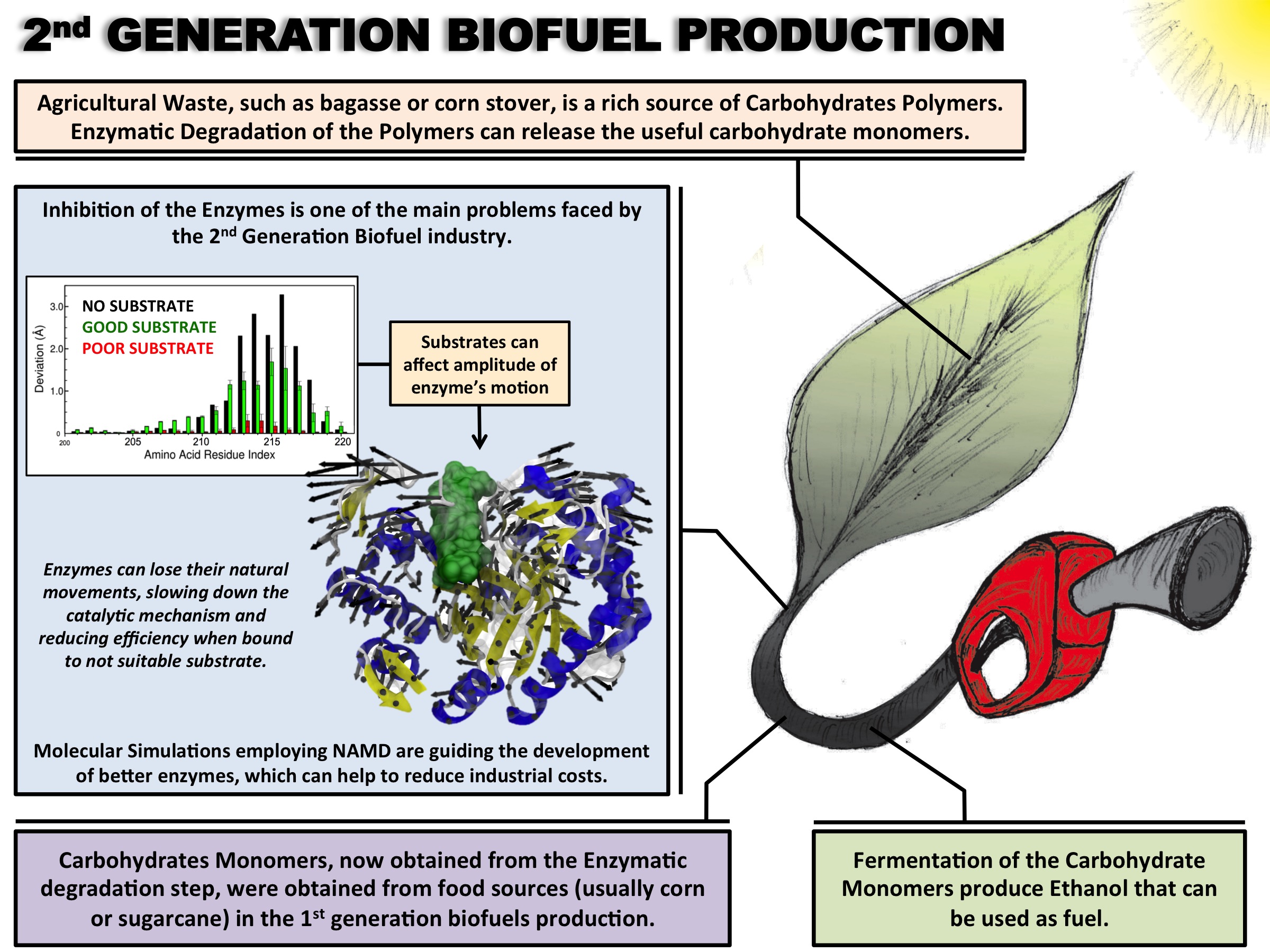
image size: 620.6KB
Biofuels are a well-known alternative to the largely used fossil-derived fuels, however the competition with food production is an ethical dilemma. Fortunately a solution is offered by second-generation biofuels, which can be produced from agricultural waste, or more specifically, from plant cell wall polysaccharides. Using the strategy of microorganisms, several enzymes are employed in the production of this advanced biofuel. However the biofuel industry faces problems such as the loss of efficiency of the enzymes that arises over time due to intermittent high concentration of non-suitable substrates. Simulations can guide biochemical experiments aimed at investigating the mechanism that makes the enzymes vulnerable to such substrates, helping the development of more efficient and, thereby, less costly enzymes. A recent study, based on molecular modeling with NAMD, reported that reduction of efficiency in an important enzyme, know as Man5B, is associated with a loss of the enzyme's flexibility. Molecular dynamis simulations showed that a poor substrate slows down a crucial opening and closing movement of the enzyme's catalytic cleft while a good substrate keeps the movement almost intact. The insight is of crucial importance since it suggests mutations to enzymes presently employed in second-generation biofuel production that need to be replaced less often and, thereby, rendering the production more cost-effective. Read more on our biofuels website.
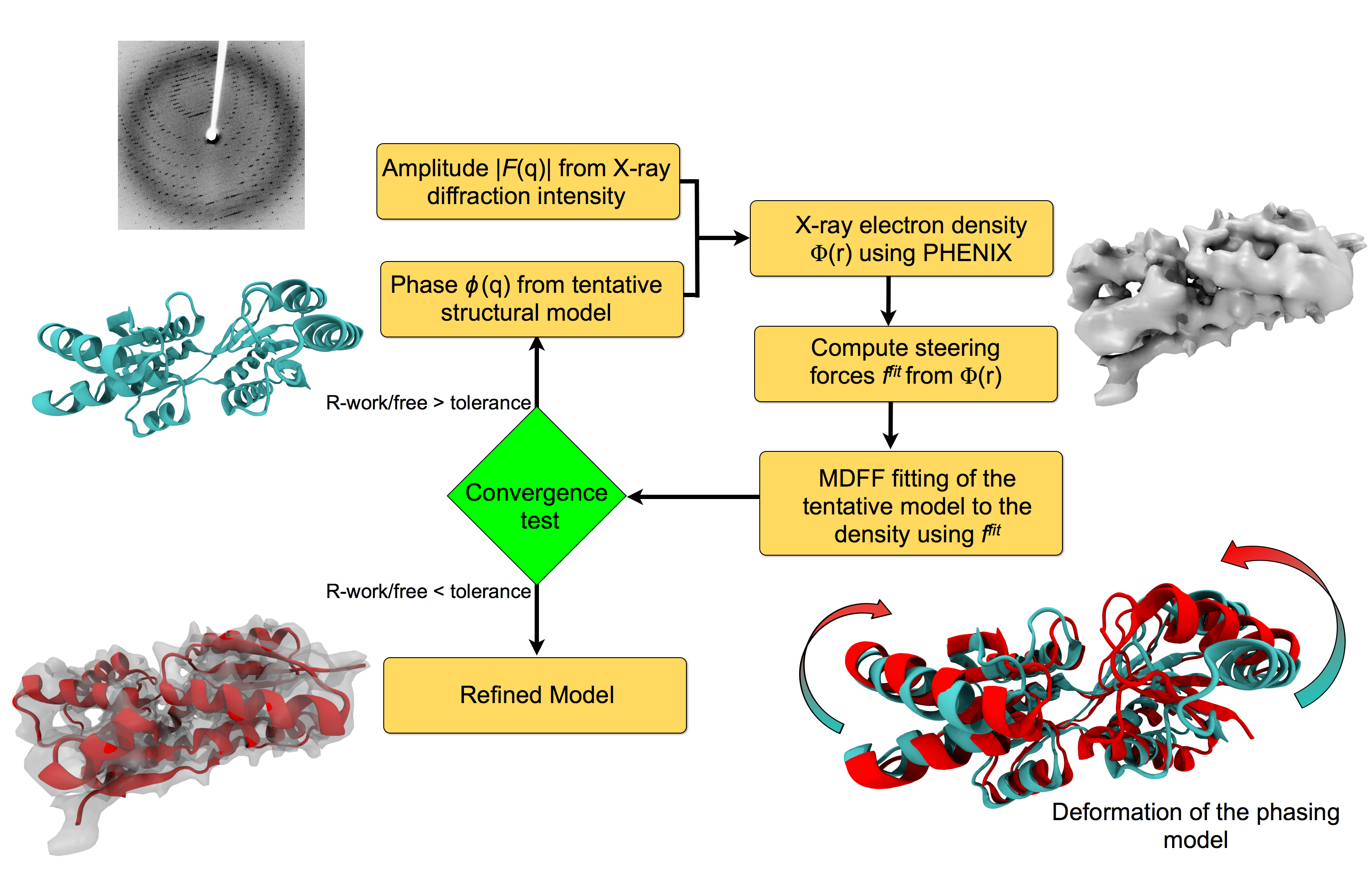
image size:
151.9KB
made with VMD
For many, the word 'X-ray' conjures up the images of white bones on black backgrounds hanging on the wall of a doctor's office. However, X-rays have played another important role for the past 100 years through their use in the determination of chemical structures at atomic level detail, starting with the first ever structure of table salt in 1924. Since then, the diffraction properties of X-rays, when shone on a crystal, have been used to solve increasingly large and complex structures including those of biological macromolecules found inside living cells. X-ray crystallography has become the most versatile and dominant technique for determining atomic structures of biomolecules, but despite its strengths, X-ray crystallography struggles in the case of large or flexible structures as well as in the case of membrane proteins, either of which diffract only at low resolutions. Because solving structures from low-resolution data is a difficult, time-consuming process, such data sets are often discarded. To face the challenges posed by low-resolution, new methods, such as xMDFF (Molecular Dynamics Flexible Fitting for X-ray Crystallography) described here, are being developed. xMDFF extends the popular MDFF software originally created for determining atomic-resolution structures from cryo-electron microscopy density maps (see the previous highlights Seeing Molecular Machines in Action, Open Sesame, Placing New Proteins, and Elusive HIV-1 Capsid). xMDFF provides a relatively easy solution to the difficult process of refining structures from low-resolution data. The method has been successfully applied to experimental data as described in a recent article where xMDFF refinement is explained in detail and its use is demonstrated. Together with electrophysiology experiments, xMDFF was also used to validate the first all-atom structure of the voltage sensing protein Ci-VSP, as also recently reported. More on our MDFF website.
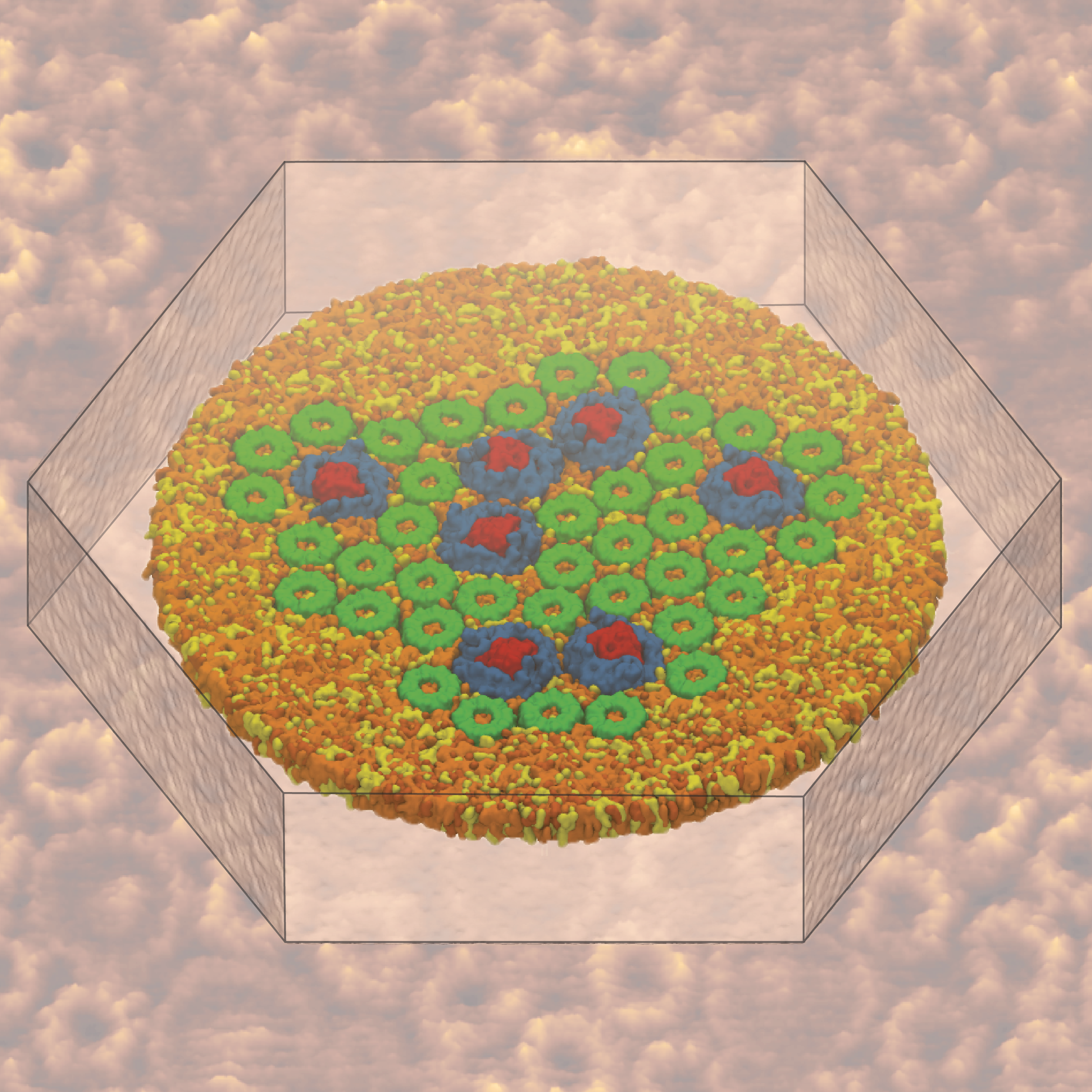
Atomic force microscopy (AFM) gives us a low-resolution glimpse of life at the nanometer scale. Now scientists can bring microscopy images to life by combining the microscopy data with atomic-detail structures to re-create the imaged system on the computer. As recently reported, Center scientists constructed an atomic-resolution model of a photosynthetic membrane based on AFM data showing the locations of the many light-harvesting proteins that inhabit the membrane. After using NAMD on petascale computers, like Blue Waters and Titan, to relax the 20-million atom membrane, Center scientists used the model to study the migration of energy among the light-harvesting complexes, as well as the mobility of quinone molecules in the membrane. The photosynthetic membrane patch was found to have a very high (90%) light-harvesting efficiency; further, it was found that the light-harvesting proteins could be considerably less tightly packed in the membrane with minimal loss of efficiency. Read more on our website.
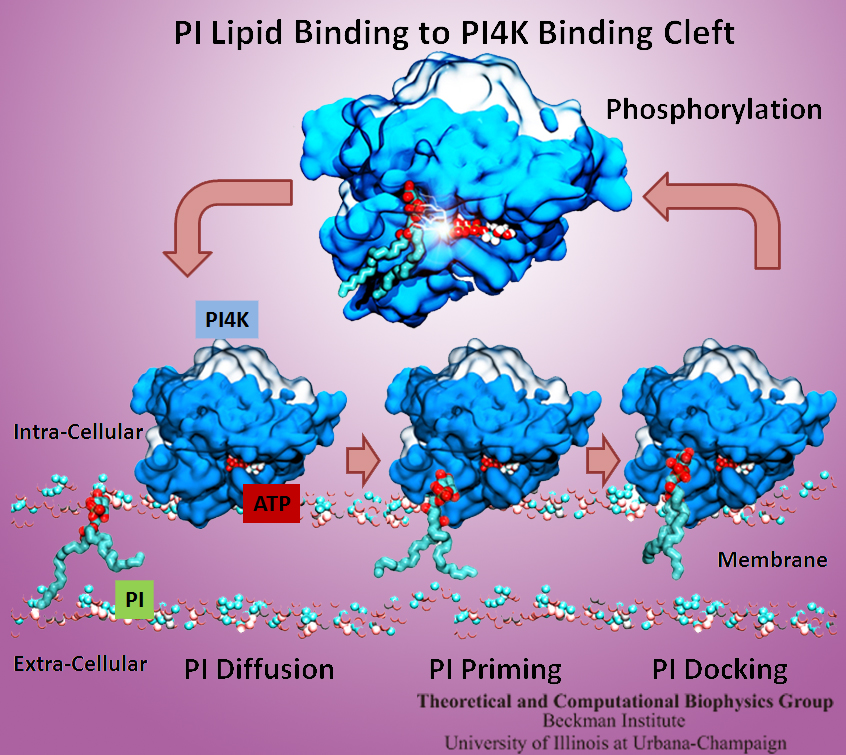
The Golgi apparatus found in so-called eukaryotic cells acts like Amazon.com, namely accepting delivery of newly synthesized proteins, packaging them, and sending them out. However, in comparison to Amazon.com the Golgi apparatus uses immensely more advanced packaging materials made of a multitude of lipids. The various lipids form membranes in the shape of vesicles. Depending on lipid type specific goods are packaged inside the vesicles, specific locations in the cell receive the packages, content is emptied there and packages are retrieved. To achieve the series of steps just outlined, lipids as the main actors need to be coordinated. One way is to recognize lipids forming vesicle membranes and to modify them to be readied for a subsequent step, for example going from release step to retrieval step. For this purpose eukaryotic cells engage a special class of proteins, named kinases, that can recognize membrane lipids and phosphorylate them, adding a so-called phosphate group. As reported recently, a team of experimental and computational scientists determined the atomic structure of a key member in the kinase family, phosphatidylinositol 4-kinase (PI4K). The scientists discovered not only the structure, but also how PI4K captures and phosphorylates a particular type of lipid molecule, thereby changing a vesicular membrane and turning on the next step in the cellular package delivery system. The discoveries, made possible through the software NAMD and VMD, are expected to have an impact on the design of novel drugs that suppress cancer cell growth. More on our kinase website.
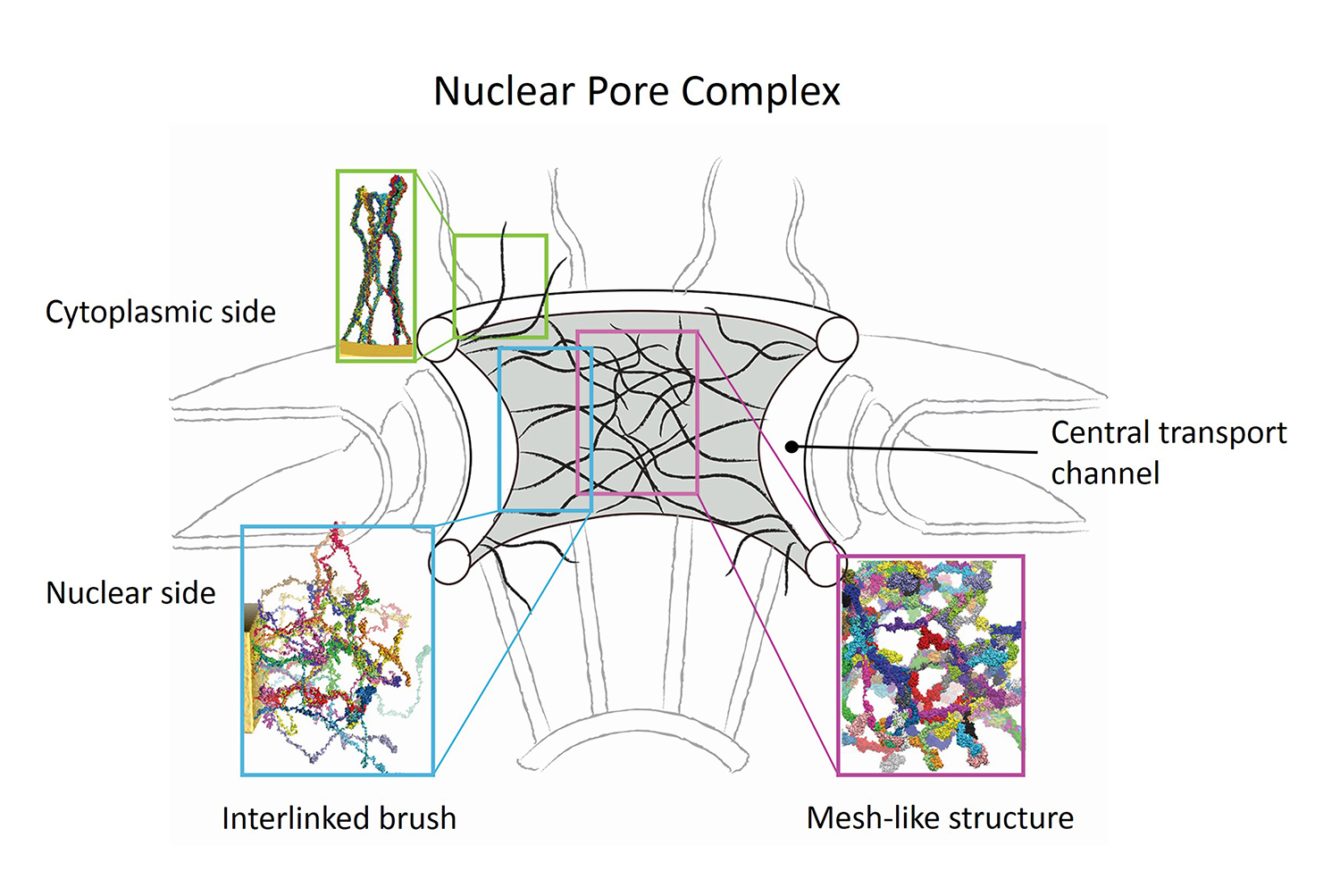
image size:
579.0KB
made with VMD
Nanoengineers building nanodevices achieve technological solutions at scales of 100 nanometers or 0.0001 mm. Nanoengineering is a brand new human technology, just a few decades old. In living cells, nanoengineering solutions are actually a few billion years old and therefore much more intricate. An impressive example is the nuclear pore, hundreds to thousands of which dot the nuclear membrane that separates in eukaryotic cells the genome and its molecular control factors from the cytoplasm of the cell. Only since very recently could cell biologists begin to resolve the molecular architecture of the nuclear pore. Given the pore's many-fold functions, like letting small molecules pass easily, but larger ones only as cargoes of special proteins, the transport factors, or adapting the pore size when large cargoes need to pass, the architecture of the nuclear pore is complex, involving an assembly of hundreds of proteins. The interior of the pore is filled with 600 amino acid-long "finger" proteins tethered at the periphery. The finger proteins are largely disordered such that experimental methods lack resolving power and computational modeling is needed to figure out their dynamic arrangement and traffic control function, but such modeling was largely unfeasible; only a small fraction of the nuclear pore volume could be covered computationally. The advent of petascale computing increased the size-scale of biomolecular simulations hundred-fold and a recent report employing the programs NAMD and VMD took advantage of the new generation of computers, simulating the dynamic, disordered arrangement of nuclear pore proteins. The simulations, still at an early stage, suggest a detailed, atomic level picture of the nuclear pore interior together with an explanation of molecular traffic control. More on our nuclear pore website.
image size:
199.7KB
made with VMD
Our body uses several defense mechanisms against seasonal flu, the common affliction caused by influenza viruses. By taking a yearly flu shot, our body's defense based on antibodies is trained and envoked. A defense system not based on antibodies acts at the very front line of influenza virus attack, namely the lungs. For this protection the body uses so-called lung surfactant proteins that coat the inner lining of the lungs to keep a wet film on the lung surface needed for oxygen-carbon dioxide exchange. The lung surfactant proteins also serve as police against influenza viruses. For this purpose the lung surfactant protein D (SP-D) recognizes a protein component of the virus surface, namely hemagglutinin, and handcuffs the sugar molecules bound to hemagglutinin. A previous experimental-simulation study (see October 2012 highlight) found that SP-D of pigs exhibits a stronger inhibitory activity against influenza A virus in this regard than does human SP-D. In a recent study, researchers have now boosted the protective ability of human SP-D by introducing mutations. Molecular dynamics simulations using NAMD suggest that the mutated human SP-D employs a different and stronger blocking mechanism on the active site of influenza A virus than native SP-D does. Combined with experimental results, the simulations suggest a mechanism through which SP-D acts, namely, by handcuffing viruses together and, thereby, preventing viral entry into cells. The findings from this research might lead to a new protection against seasonal flu, namely a nasal spray containing mutated lung surfactant proteins that strengthen a person's armada of defense proteins on the lung surface. More on our lung surfactant protein website.
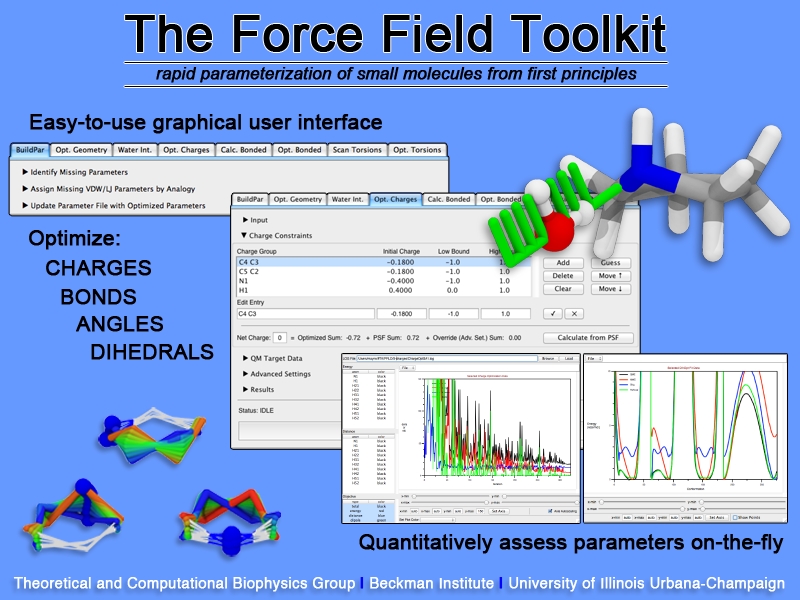
image size:
307.6KB
Structural biologists are increasingly turning to simulation methods to investigate the connections between molecular structure and biological function. Classical molecular dynamics (MD) simulations, such as those performed by the simulation software NAMD, rely on potential energy functions requiring parameters to describe atomic interactions within the molecular system. While these parameters are available for the most commonly simulated biopolymers (e.g., proteins, nucleic acids, carbohydrates), many small molecules and other chemical species lack adequate descriptions. The complexity of developing these parameters severly restricts the application of MD technologies across many fields, including most notably drug discovery. Recently, researchers have developed software, the Force Field Toolkit (ffTK), that greatly reduces these limitations by facilitating the development of parameters directly from first principles. ffTK, distributed as a plugin for the molecular modeling softare VMD, addresses both theoretical and practical aspects of parameterization by automating tedious and error-prone steps, performing multidimensional optimizations, and providing quantitative assessment of parameter performance--all from within an easy-to-use graphical user interface. Additional information on ffTK, including documentation and screencast tutorials, can be found here.


