MDFF Spotlights
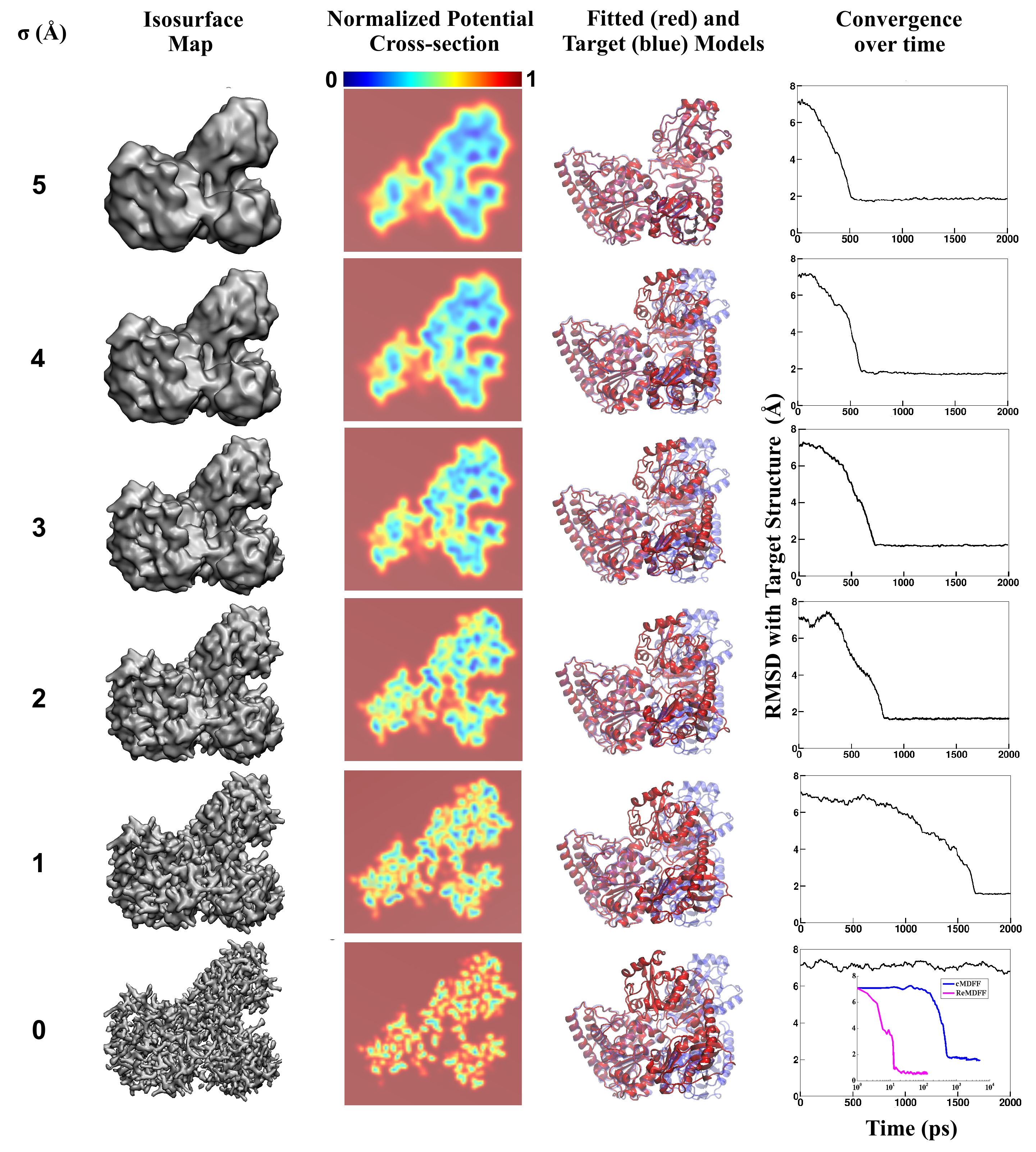
image size:
3.3MB
made with VMD
Living cells are brimming with the activity of macromolecular complexes carrying out their assigned tasks. Structures of these complexes can be resolved with cryo-electron microscopy (cryo-EM), wherein the complexes are first freeze-shocked into states characterizing their action and subsequently imaged by detection cameras. Recent advances in direct detection camera technology enable today's cryo-EM laboratories to image the macromolecular complexes at high-resolution, giving us a better view of the cell than ever before. Computational techniques like molecular dynamics flexible fitting (MDFF) are a key tool for producing atomic models of the imaged molecules, providing greater insight into their structure and function. The increased resolution of EM maps, which contain sharp valleys capable of trapping structures, presents a challenge to MDFF which was originally developed for maps in a lower resolution range. However, a recent study unveils two new techniques called cascade (cMDFF) and resolution exchange (ReMDFF) molecular dynamics flexible fitting to overcome the hurdles posed by high-resolution maps. The refinement is achieved by interpreting a range of cryo-EM images, starting with an image of fuzzy resolution and progressively improving the image's contrast until near-atomic resolution is reached. These techniques were employed to solve the structure of the proteasome, the recycling machine of the human cell. New analysis schemes that look at the flexibility of the obtained structure provide a measure of model uncertainty within the near-atomic EM images, improving their contrast. All the tools are available on cloud computing platforms allowing community-wide usage at low monetary cost; the complex computations can now be performed at the cost of a cup of coffee.
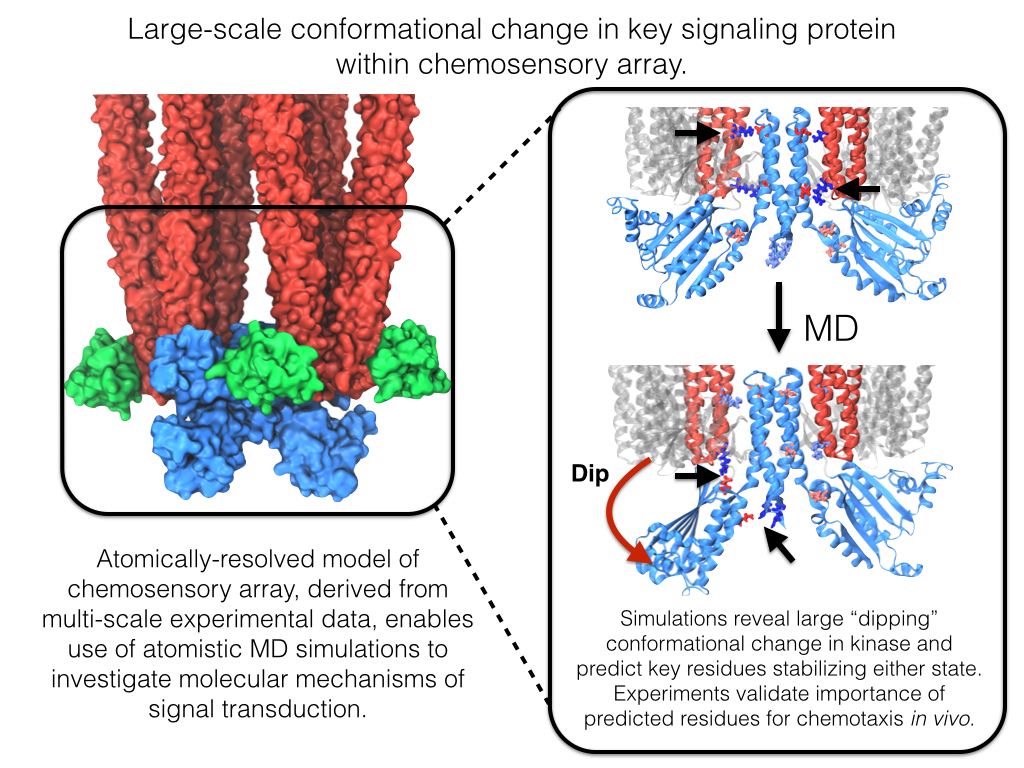
image size:
642.1KB
made with VMD
Motile bacteria position themselves within their habitats optimally, seeking proximity to favorable growth conditions while avoiding unfavorable ones. Cues used for this placement come in the form of small chemicals, so-called attractors and repellants, as well as physical factors such as favorable visible light and unfavorable UV radiation. To balance such a broad range of factors, bacteria monitor their environments and respond by way of a fundamental sensory capability known as chemotaxis. Chemotactic responses in bacteria involve large complexes of sensory proteins, known as chemosensory arrays, that process the information obtained from the bacteria's habitat to determine its swimming pattern. In this sense, the chemosensory array functions as a bacterial brain, transforming sensory input into motile output. Despite great strides in the understanding of how the chemosensory array's constituent proteins fit and work together, a high-resolution description of the kind needed to explore in detail the molecular mechanisms underlying sensory signal transduction within the array has remained elusive. A new study, utilizing cryo-electron microscopy and molecular dynamics simulations with NAMD, reports the highest resolution images yet of the bacterial brain's molecular anatomy. Using computational techniques, structural data from X-ray crystallography and electron microscopy are compared to derive an atomically resolved model of the chemosensory array's extended molecular structure that involves millions of atoms. Subsequent simulations of the model revealed a novel conformational change in a key sensory protein, that is interpreted as a key signaling event in the translation of chemosensory information into swimming pattern. More details on this work can be found in a recent news release as well as on our bacterial chemotaxis website.
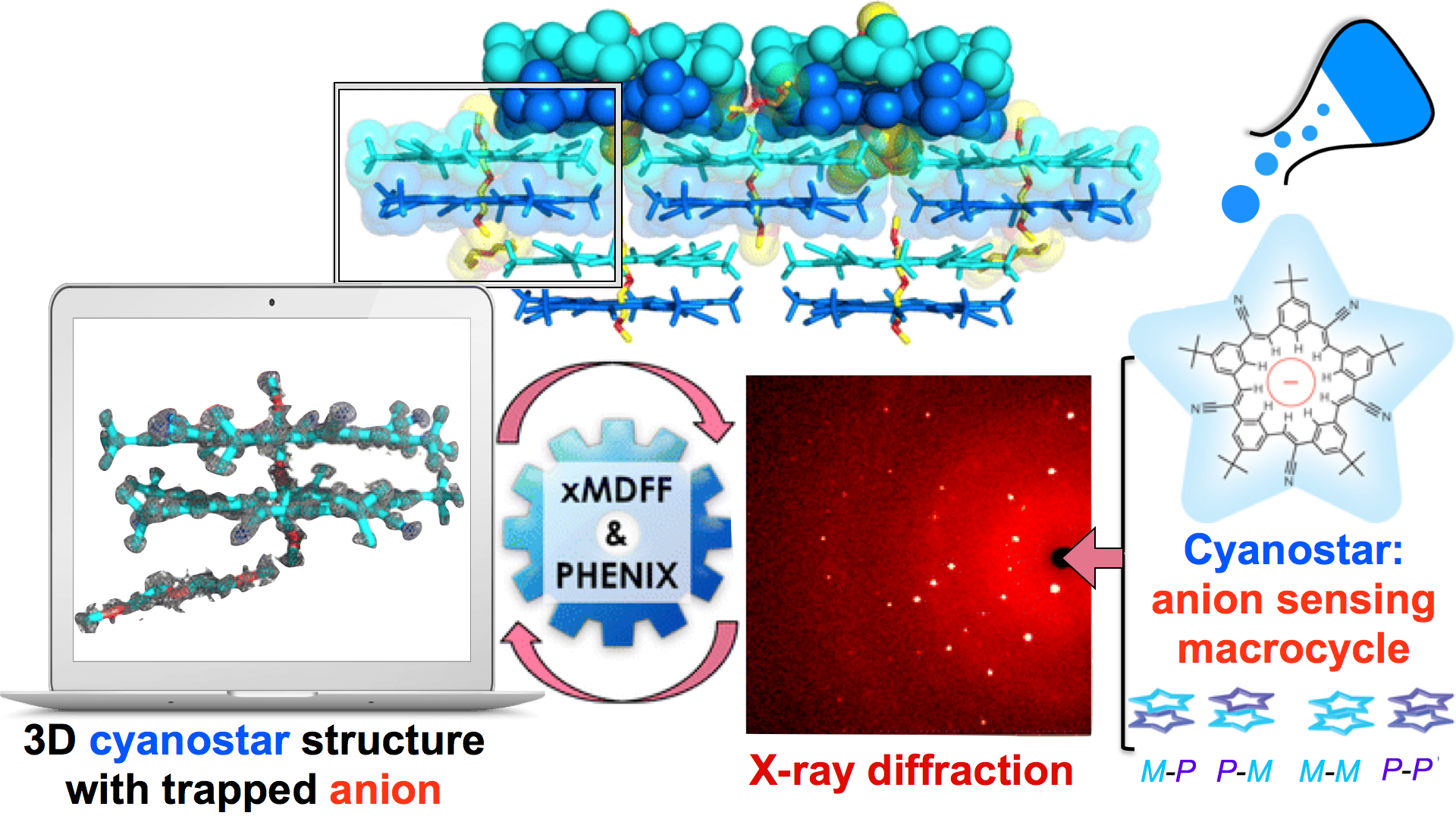
image size: 1.3MB
Through crystallography researchers have solved the structures of many molecules, chemists those of small organic and inorganic molecules produced via chemical synthesis and biologists those of large macromolecules as they arise in living cells. But the borderline between small and large is not well defined and indeed chemical synthesis produces today supramolecules, namely, coordination cages, abiological foldamers, metal-organic frameworks and covalent-organic frameworks, that are of similar size and disorder as biological macromolecules, yet, elicit extremely intricate electronic properties resembling the small-molecules. To image the supramolecules chemists and biologists have now joined forces as reported recently . Key to their strategy are methods developed earlier by computational structural biologists, xMDFF (see August 2014 highlight xMDFF Enhances X-Ray Structures) and PHENIX , and by biomolecular modelers, NAMD (see Jan 2015 highlight NAMD 2.10 Advances Simulations Large and Small ) and ffTK (see Sep 2013 highlight May the Force be With You ), that were imported to chemistry. The team focussed on the structural description of the molecule cyanostar, an anion-sensing organic macrocycle. Two cyanostar molecules forming a pair were resolved atom-by-atom in two distinct stereoisomeric forms, at an extremely high resolution of 0.84 Å, with flexible solvent molecules sitting in their macrocyclic and intermolecular void spaces. More on our MDFF website.
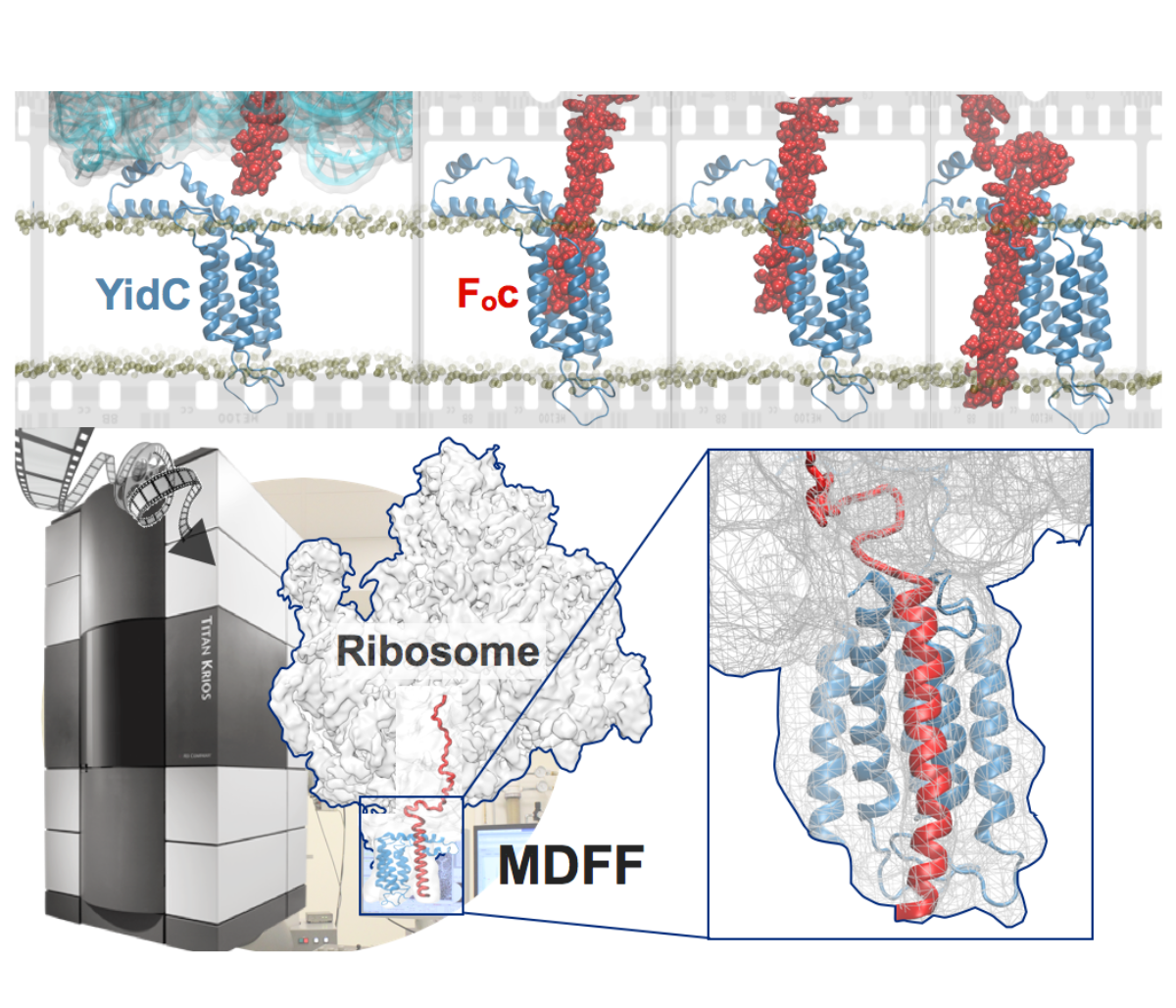
image size:
901.7KB
made with VMD
Synthesis and placement of new proteins in a living cell poses a challenge for the cellular machinery, in particular in case of so-called membrane proteins. Starting with nothing more than a sequence of DNA, the cell has to translate the genetic code, stitch together the constituent amino acids, and then place the newly made protein where its function is needed, namely the cell membrane. To meet the challenge the cell employs a molecular machine for the synthesis of proteins, the ribosome (see the Dec. 2009 highlight on Managing the Protein Assembly Line ), as well as special proteins that translocate newly synthesized proteins out of the machine into the cell membrane (see the Feb. 2011 highlight on Placing New Proteins ). Depending on the complexity of the membrane protein insertion, different protein systems are used for the translocation, in most cases the systems involving complexes of several, even many, proteins. Now however, the structure and function of the simplest translocating protein system has been solved, which actually is made of only a single protein, called YidC. Despite its simplicity, structure determination of YidC was difficult, taking three decades. Successful structure determination was recently reported here, and involved the combination of cryo-electron microscopy, mutational experiments and computer simulations, the latter using NAMD, VMD and MDFF. The structure that was discovered shows a distinctive arrangement of five trans-membrane helices and reveals how a single copy of YidC interacts with the ribosome at the ribosomal tunnel exit and identifies a site for membrane protein insertion at the YidC protein-membrane lipid interface. The quality of this atomic model is validated by its close agreement with a recently published crystal structure of E. Coli YidC (here). More on our protein translocation website.
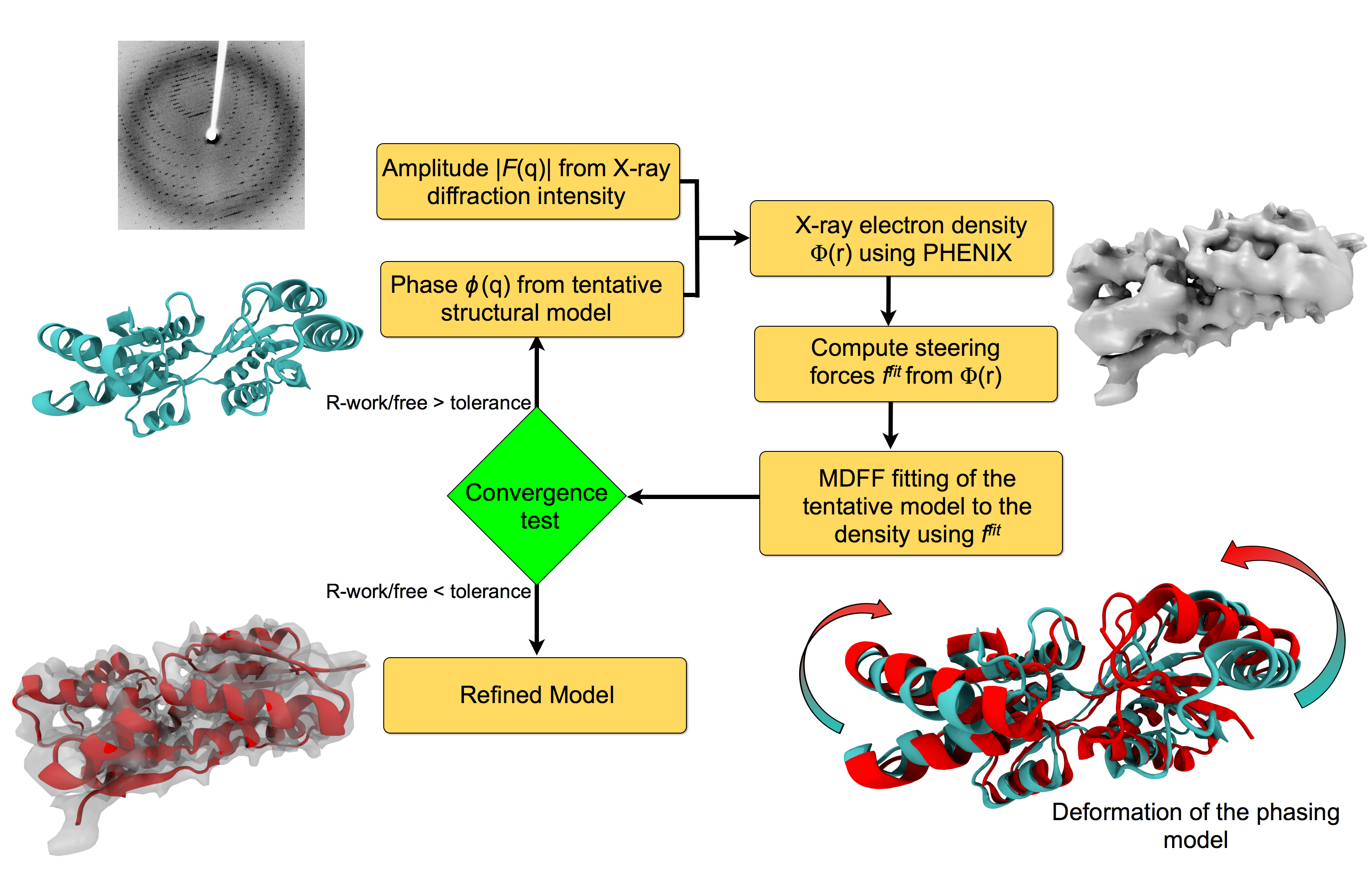
image size:
151.9KB
made with VMD
For many, the word 'X-ray' conjures up the images of white bones on black backgrounds hanging on the wall of a doctor's office. However, X-rays have played another important role for the past 100 years through their use in the determination of chemical structures at atomic level detail, starting with the first ever structure of table salt in 1924. Since then, the diffraction properties of X-rays, when shone on a crystal, have been used to solve increasingly large and complex structures including those of biological macromolecules found inside living cells. X-ray crystallography has become the most versatile and dominant technique for determining atomic structures of biomolecules, but despite its strengths, X-ray crystallography struggles in the case of large or flexible structures as well as in the case of membrane proteins, either of which diffract only at low resolutions. Because solving structures from low-resolution data is a difficult, time-consuming process, such data sets are often discarded. To face the challenges posed by low-resolution, new methods, such as xMDFF (Molecular Dynamics Flexible Fitting for X-ray Crystallography) described here, are being developed. xMDFF extends the popular MDFF software originally created for determining atomic-resolution structures from cryo-electron microscopy density maps (see the previous highlights Seeing Molecular Machines in Action, Open Sesame, Placing New Proteins, and Elusive HIV-1 Capsid). xMDFF provides a relatively easy solution to the difficult process of refining structures from low-resolution data. The method has been successfully applied to experimental data as described in a recent article where xMDFF refinement is explained in detail and its use is demonstrated. Together with electrophysiology experiments, xMDFF was also used to validate the first all-atom structure of the voltage sensing protein Ci-VSP, as also recently reported. More on our MDFF website.
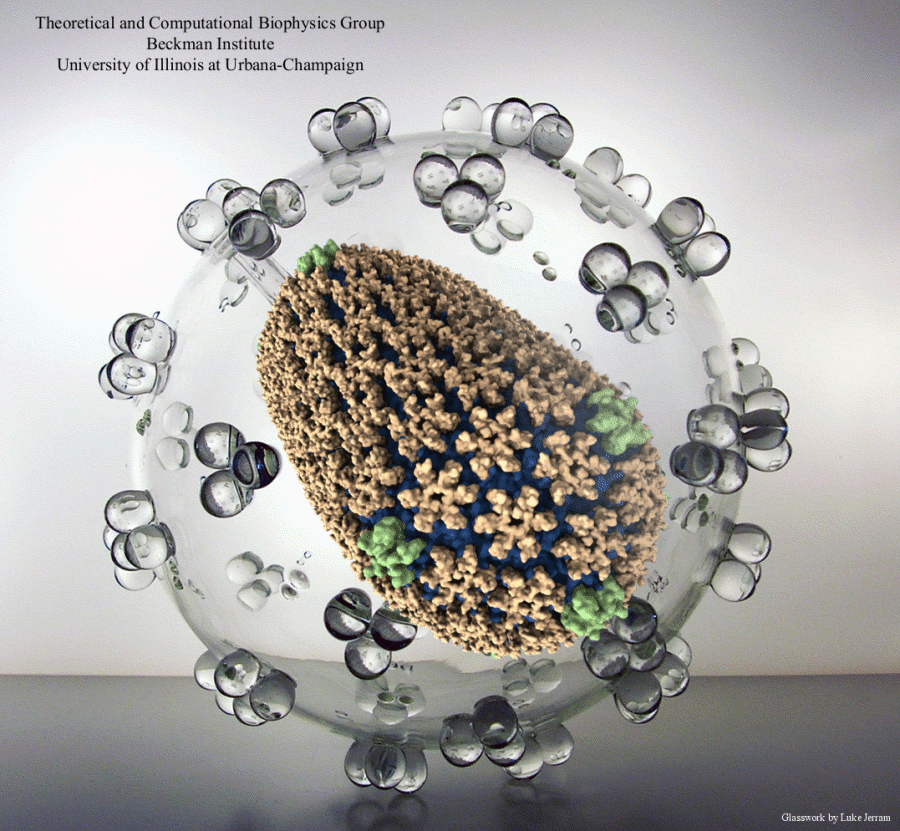
Human immunodeficiency virus type 1 (HIV-1) is the major cause of AIDS, for which treatments need to be developed continuously as the virus becomes quickly resistant to new drugs. When the virus infects a human cell it releases into the cell its capsid, a closed, stable container protecting the viral genetic material. However, interaction with the cell triggers at some point an instability of the capsid, leading to a well timed release of the genetic material that merges then with the cell's genes and begins to control the cell. The dual role of the capsid, to be functionally both stable and unstable, makes it in principle an ideal target for antiviral drugs and, in fact, treatments of other viral infections successfully target the respective capsids. The size of the HIV-1 capsid (about 1,300 proteins), and its irregular shape had prevented so far the resolution of a full capsid atomic-level structure. However, in a tour de force effort, groups of experimental and computational scientists have now resolved the capsid's chemical structure (deposited to the protein data bank under the accession codes 3J3Q and 3J3Y). As reported recently (see also journal cover), the researchers combined NMR structure analysis, electron microscopy and data-guided molecular dynamics simulations utilizing VMD to prepare and analyze simulations performed using NAMD on one of the most powerful computers worldwide, Blue Waters, to obtain and characterize the HIV-1 capsid. The discovery can guide now the design of novel drugs for enhanced antiviral therapy. More information is available on our virus website, in video, and in a press release.

Rabbit hemorrhagic disease is extremely contagious and associated with liver necrosis, hemorrhaging, and high mortality in adult rabbits. First described in China in 1984, within a few years, rabbit hemorrhagic disease spread to large parts of the world and today threatens the rabbit industry and related ecology. The disease is caused by a virus, aptly named rabbit hemorrhagic disease virus. As reported recently, a group of experimental and computational researchers combining crystallography, electron microscopy and data-guided molecular dynamics simulations utilizing NAMD determined an atomic model of the capsid, namely the protein shell that surrounds the genetic material of the virus. The capsid simulations involved 10 million atoms and have become feasible only through Blue Waters, a brand new petascale supercomputer. The atomic model, analyzed by means of VMD, recently adapted to studies of very large structures, resolves the structural framework that furnishes both mechanical protection to the viral genes as well as a quick release mechanism after a virus enters a host cell. Researchers can use the detailed knowledge of the capsid structure to develop vaccines against rabbit hemorrhagic disease. More information is available on our virus website and in a news story.
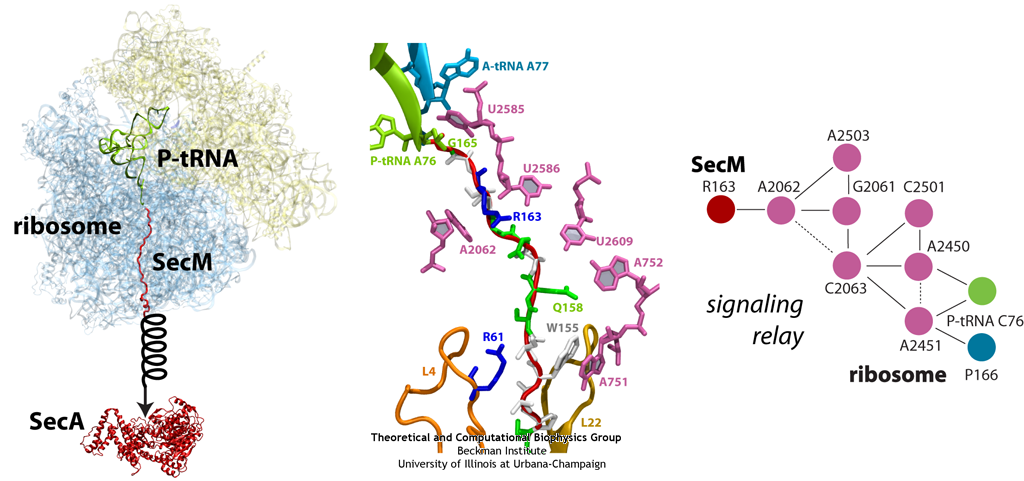
image size:
299.9KB
made with VMD
The ribosome functions as a cellular protein factory, synthesizing practically all the proteins in the cell based on blueprints read from DNA (see the April 2012 highlight and Dec. 2009 highlight). However, unlike an assembly line, the ribosome has no foreman directing it. Instead, regulation of protein synthesis is managed by a number of external, and internal, signals. For example, the protein TnaC halts its own synthesis in the ribosome to promote that of another protein (see the May 2010 highlight). Similarly, synthesis of the protein SecA, a translocase that aids in pushing newly made proteins across membranes, is controlled through the nascent protein SecM. Regulation of SecA levels is the only function of SecM, which is degraded as soon as it leaves the ribosome. It is the stalling of one ribosome by SecM that provides enough time for secA, which resides on the same messenger RNA as secM, to be translated by a second ribosome, thus upregulating SecA production. When enough SecA has been produced, it pulls on the portion of SecM outside the ribosome, relieving its stalled state. The critical interactions that cause stalling have now been identified through a combination of molecular dynamics and cryo-electron microscopy via MDFF and NAMD. As recently reported, these interactions form a relay connecting SecM in the exit tunnel to the ribosome's key catalytic center, preventing synthesis and thus explaining how SecM stalls inside "its" ribosome. Additionally, steered MD simulations revealed how SecA can cause the nascent SecM to become unstuck, by breaking those same interactions. More details are provided on our ribosome website.
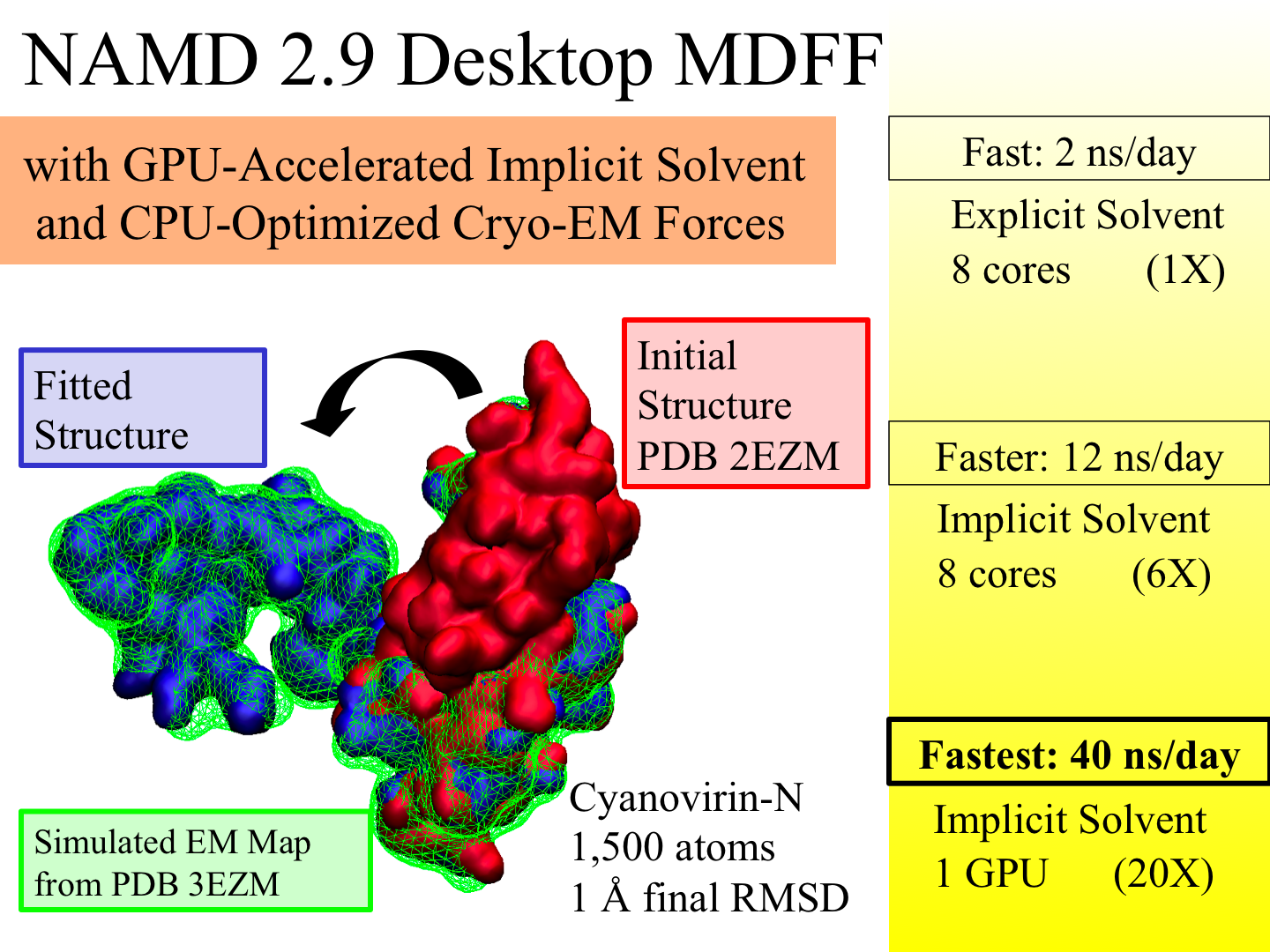
image size:
883.7KB
made with VMD
X-ray crystallography resolves the structures of the molecular machines in living cells at an atomic level of detail, but only in states that can be captured as crystals, which are often not functional states. Cryo-electron microscopy enables a more complete view of biomolecular conformational variability, but at lower resolution. The molecular dynamics flexible fitting (MDFF) method combines the atomic detail of crystallographic structures with lower-resolution cryo-electron microscopy to synthesize all-atom models of complex macromolecular aggregates such as the ribosome in multiple functional states. The 2.9 release of NAMD combines GPU acceleration of implicit solvent simulation, optimizations exploiting shared memory within a single machine, and a faster "lite" grid forces implementation to bring MDFF capability from the supercomputer to the desktop. VMD can connect to a running simulation to visually monitor the progress of the simulation or to interactively steer a molecule with either the mouse or a haptic (force-feedback) interface device. The convergence of methodology, software, and hardware advances thus opens what was once the domain of extremely expensive equipment to commodity computers.

made with VMD
Viruses reproduce by splicing their genetic material into a host cell, causing the cell to manufacture new viruses. This genetic material is protected outside of the host cell by a protein capsid, which disassembles inside a new cell to complete the infection process. Simulation of viral infection has progressed significantly since the first all-atom virus simulation was done with NAMD in 2006 and is one of the driving biomedical projects for the software. A new collaboration with the Pittsburgh Center for HIV Protein Interactions has applied molecular dynamics flexible fitting to construct the first all-atom structure of an HIV virus capsid in its tubular form (shown). This structure is now being simulated as one of six early science projects on the Blue Waters petascale supercomputer being installed at Illinois. These large-scale simulations are enabled by the 2.9 release of NAMD, which includes a new high-performance interface to the Cray Gemini network of Blue Waters. Smaller simulations may also leverage petascale computing through a new replica-exchange framework that supports parallel tempering and integrates with the collective variables module for umbrella sampling conformational free energy calculations. GPU acceleration enhancements include minimization and implicit solvent support as well as exploitation of shared memory, extending performance gains to the desktop.
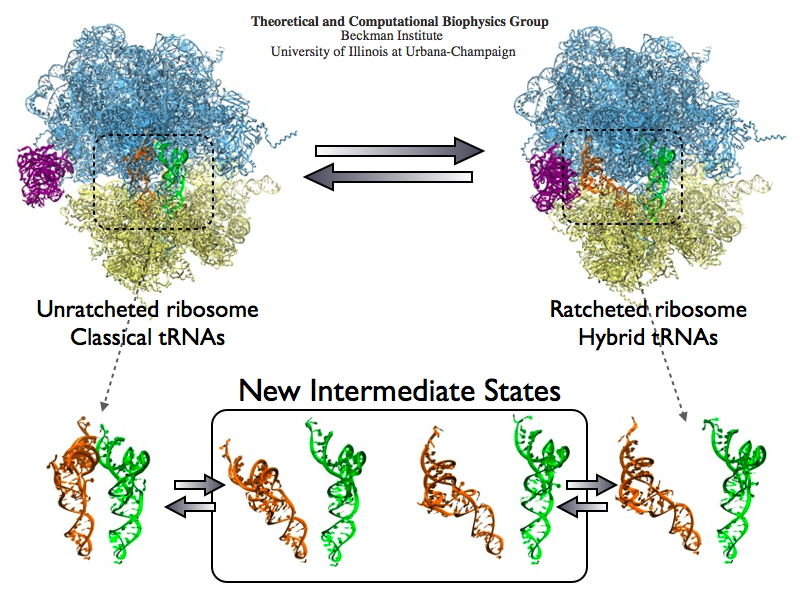
image size:
488.4KB
made with VMD
The ribosome is the protein assembly line in all living cells. The building material for new proteins is supplied by RNA molecules, called tRNA. They enter and move through the ribosome, each adding a new amino acid to the nascent proteins according to the genetic sequence provided through so-called messenger RNA. During the tRNAs' translation through the ribosome, the ribosome itself is not static either. A ratcheting motion and other large scale motions can be observed. However, the exact tRNA and ribosome motion were not clear. Using images from cryo-electron microscopy, MDFF, a computational method based on NAMD, allows one to see the moving parts within the ribosome in great detail. MDFF (see the June 2008 highlight) already provided crucial and unique insights into different aspects of protein synthesis, such as translational arrest of the ribosome by a nascent chain or translocation of an emergent protein across a membrane. In the work reported recently, MDFF revealed the presence of previously unseen intermediate states of the ribosome and its bound tRNAs during the ratcheting motion. A thorough analysis of these states pictures the ribosome as a molecular machine using Brownian motion for its function. More on our ribosome website.
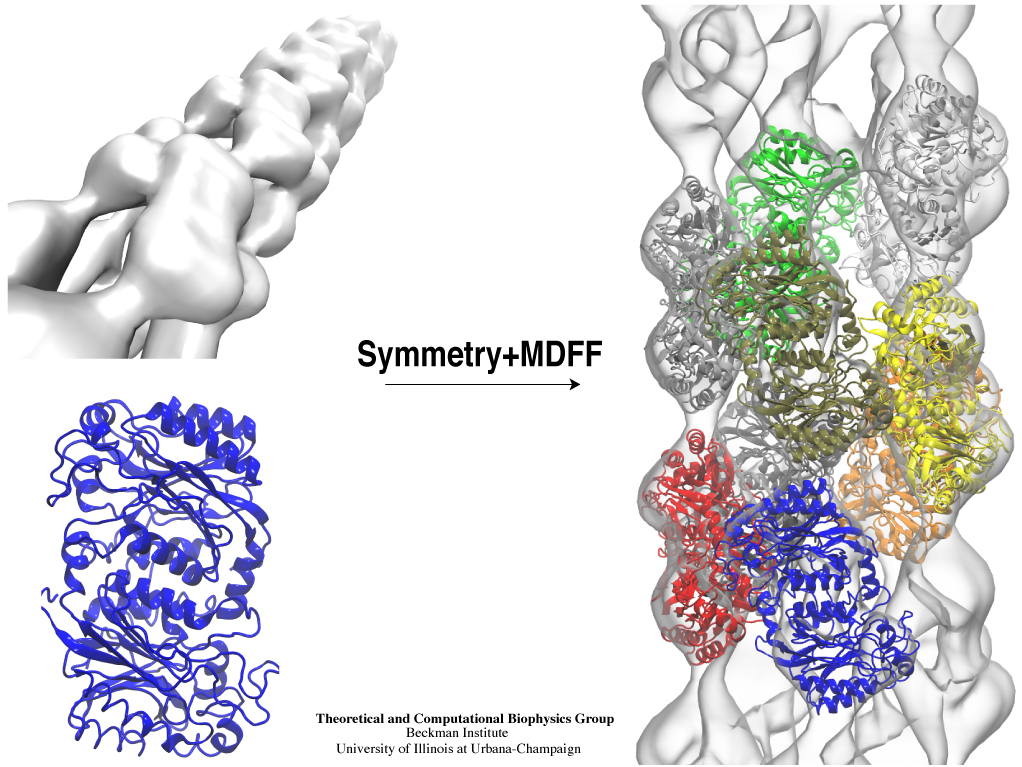
Inorganic nature brings about symmetry that one can admire, for example when one sees a polished diamond. Living nature, too, brings about symmetric structures; for example, many processes in living cells are carried out by highly symmetric protein complexes. In case of living cells symmetry comes about partially out of physical or geometrical necessity, but also partially out of its usefulness for the intended purpose. Hence, understanding symmetry in biology goes beyond related studies of the beauty of symmetry in the physical and mathematical sciences as one also asks: Does symmetry help? Put it another way: Symmetry in living cells is beautiful and useful! The proteins in symmetric complexes are often imaged by electron microscopy (EM), but unfortunately not at a resolution high enough to recognize chemical detail, as is needed in most cases, e.g., in case of the study of a symmetric multi-enzyme system. Computational biologists have developed a method to solve the resolution problem, using high resolution images obtained through X-ray scattering of related molecules and matching them through molecular dynamics using NAMD to the image seen in EM. This method, called molecular dynamics flexible fitting (MDFF) has been highlighted previously and is described on our MDFF website. As reported recently, MDFF has now been extended to determine the atomic-level structure of symmetric multi-protein systems. MDFF has been applied successfully to three highly symmetric multi-protein systems: (i) GroEL-GroES, a protein complex that assists proteins to fold properly into their native conformation; (ii) a nitrilase multi-enzyme system that converts massive amounts of molecules into forms more suitable for a bacterial cell; (iii) Mm-cpn, a protein complex supposedly involved in assisting protein folding in so-called archaebacteria. In all cases the symmetry of the protein complexes plays a key role. For more details, see the Method section of our MDFF website.
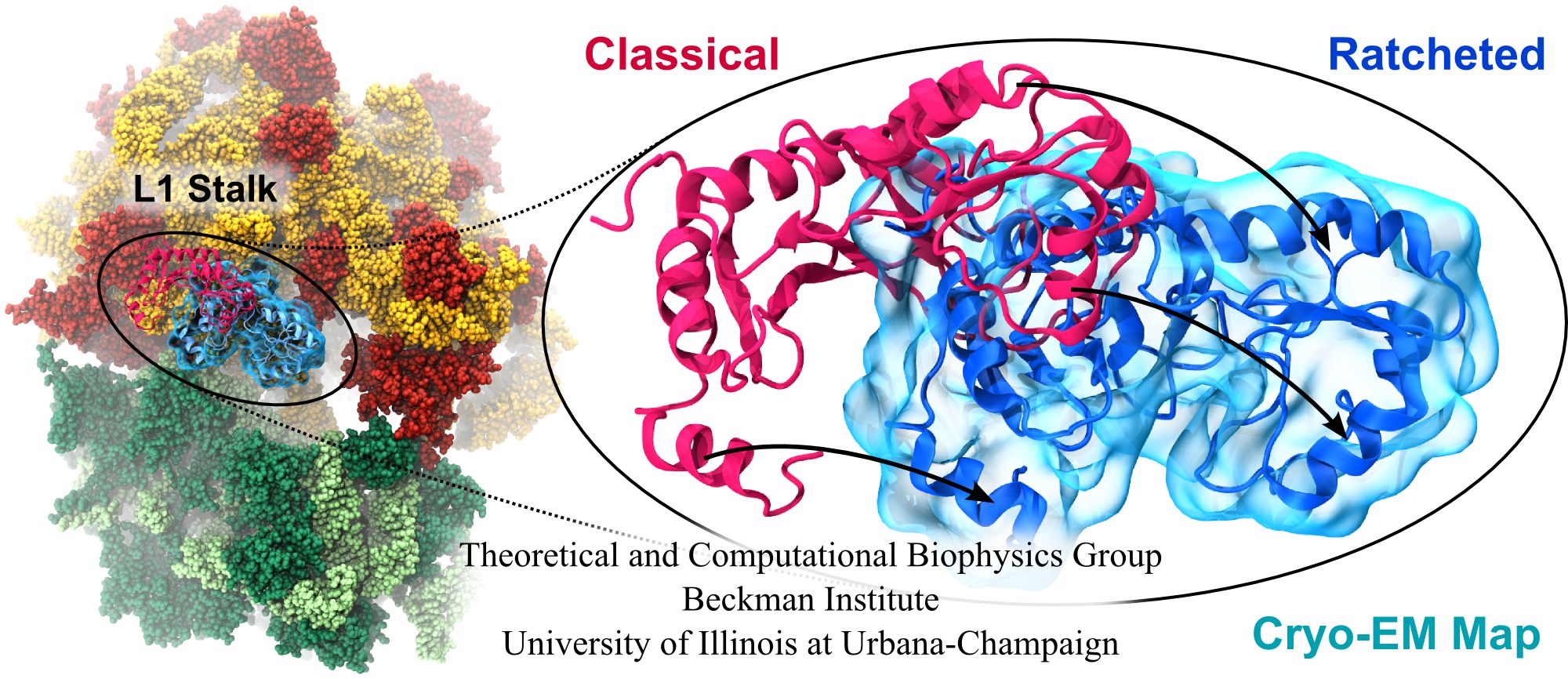
image size:
347.6KB
made with VMD
Water is the essential solvent that shapes protein structure and function, but for researchers using molecular dynamics flexible fitting (MDFF) to fit large biomolecular models to data from cryo-electron microscopy, such as fitting the classical ribosome into the ratcheted map, it was a mixed blessing. Since the network of hydrogen bonds that gives water its unique properties must rearrange as the solute moves, water molecules not only increase the size of the simulation but slow the fitting process. Leaving water out completely was a common practice, relying on the MDFF fitting potential to prevent the dehydrated protein from shriveling. The 2.8 release of NAMD provides a better option: a new implementation of the generalized Born implicit solvent model that scales to thousands of cores for large biomolecular aggregates thanks to NAMD's unique parallel structure and measurement-based load balancing system. By eliminating explicit water molecules from the simulation, an implicit solvent model helps shape protein structure while adapting immediately to new conformations. With this best-of-both-worlds option now available in NAMD, biomedical researchers using MDFF need fear water no longer.
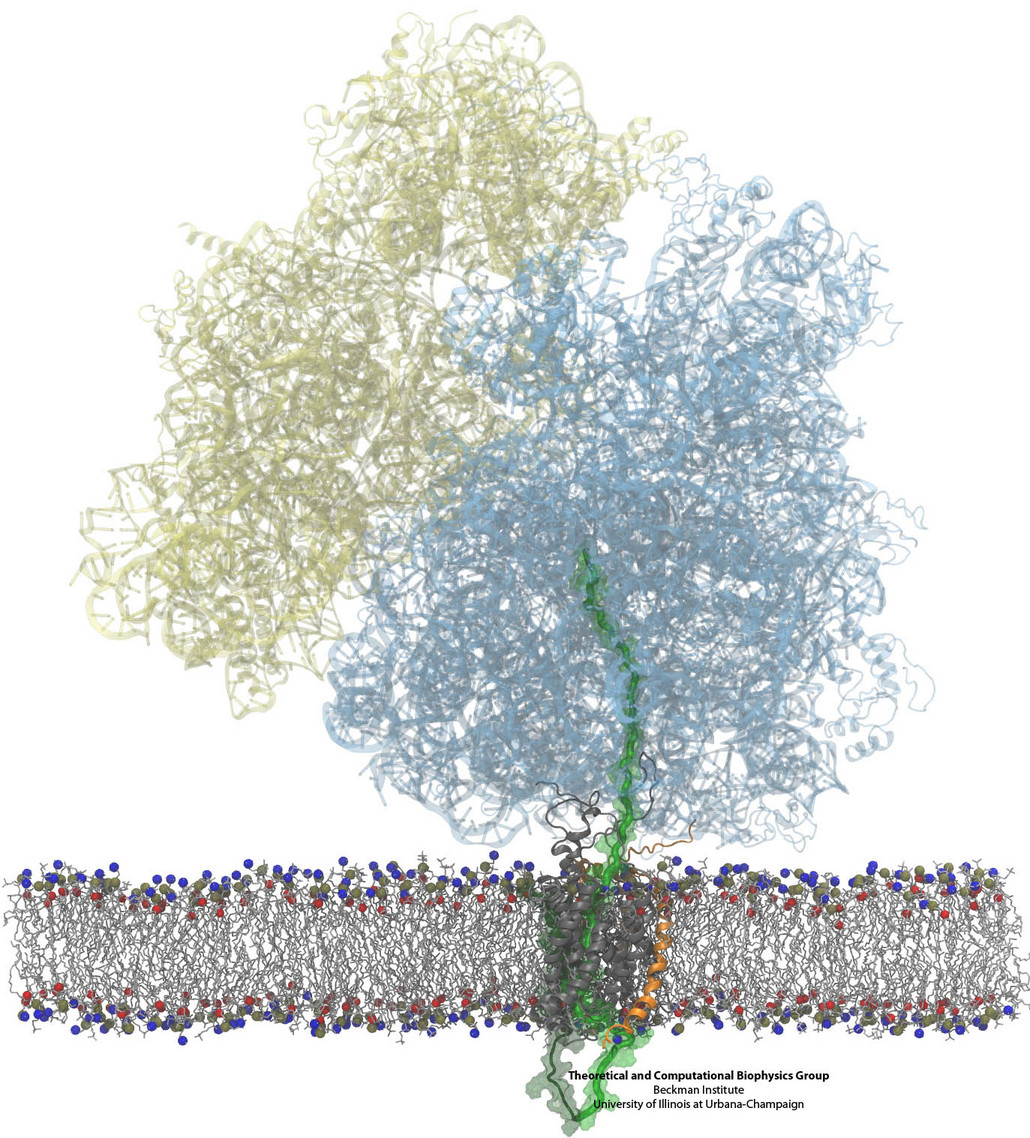
Constructing and correctly placing new proteins is a complicated task for living cells. Starting with nothing more than a sequence of DNA, the cell has to translate the genetic code, stitch together the constituent amino acids, and then place the newly made protein where its function is needed. To accomplish these feats, the cell using tools such as the ribosome, a protein factory (see the Dec. 2009 highlight Managing the Protein Assembly Line), and the protein-conducting channel, a switching station within the membrane (see the Nov. 2008 highlight Patching a Leaky Channel). All instructions for making a nascent protein and localizing it, e.g., to the watery cytoplasm or the oily membrane, are carried within its DNA sequence, but how it is read and then executed has long been unclear. Now, two recent research advances picture these processes in astonishing detail. The first advance (reported here), from a collaboration between cryo-electron microscopists and computational biologists using MDFF, shows an atomic level structure that caught the ribosome in the act of inserting a protein into a membrane. The structure reveals the newly forming protein transiting from within the ribosome to the channel and then through an open gate in the protein-conducting channel into the membrane. The second advance (reported here), accomplished with the simulation program NAMD, explained how the ribosome and the protein-conducting channel manage to pay the energy price of inserting a new protein, one amino acid a time, into the membrane. For more information see our protein translocation website.
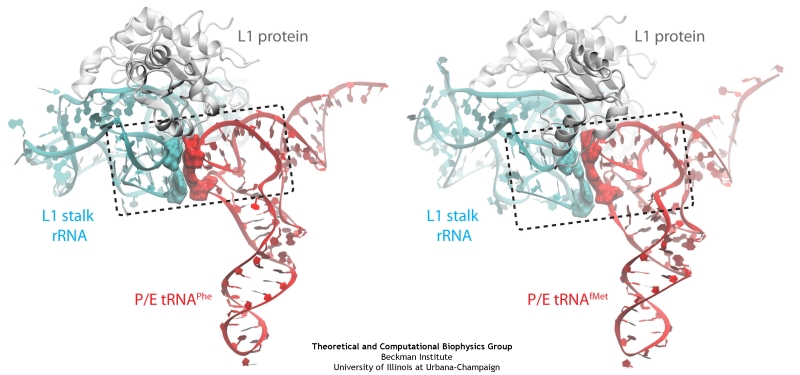
image size:
140.2KB
made with VMD
The ribosome is a molecular machine ubiquitous in all living cells and translates genetic information into proteins. Proteins are made of twenty different amino acids, strung in a linear sequence. The amino acids are coded for by the genes in DNA, but for the purpose of protein synthesis genes are transcribed into a working copy, a messenger RNA. The latter is translated by the ribosome into proteins with the help of transfer RNAs, which bring the individual amino acids. There is a transfer RNA for each of the twenty amino acids. Much progress has been made regarding the static structure of the ribosome, transfer RNA, and nascent protein components (see also the Dec 2009 and Jan 2009 highlights Managing the Protein Assembly Line and Open Sesame). Now researchers are looking into the inner workings of the whole system combining various experiments and computational modeling using NAMD and VMD. The combination yielded unprecedented detailed views of the ribosome in action as reported recently, namely how a dynamic part of the ribosome helps guiding transfer RNAs on their way out of the ribosome, and explains why transfer RNAs behave differently on their journey, depending on if they start synthesis of a protein or if they elongate a protein. More on our ribosome website.
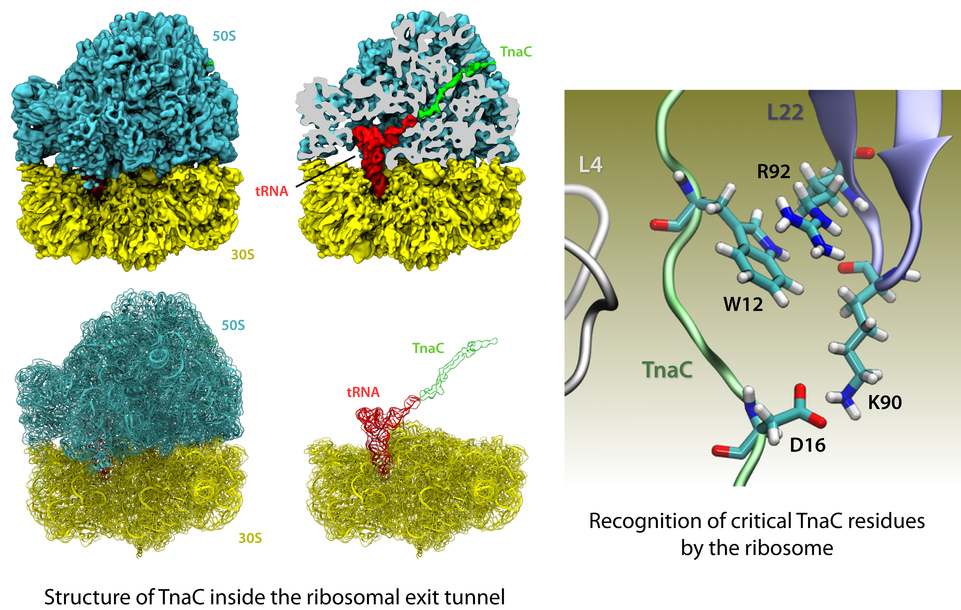
image size: 541.6KB
The ribosome is the protein factory of all cells. While proteins are being synthesized, they must travel through a tunnel inside the ribosome before they reach their required location in the cell. This tunnel recognizes certain sequences of nascent proteins and responds to them in various ways. For example, certain protein sequences recognized in the tunnel can shut the ribosome down, stopping protein synthesis. A much studied example is the bacterial protein TnaC, which by shutting down its own synthesis turns on the synthesis of proteins involved in the degradation of a molecule called tryptophan. The structure of TnaC inside the ribosome was previously determined through crystallography, electron microscopy, and computer modeling (see the December 2009 highlight on Managing the Protein Assembly Line). Simulation of TnaC dynamics inside the ribosome using NAMD and VMD has now revealed the mechanism by which TnaC shuts down the ribosome as reported recently. Answered was also the question of how the ribosome recognizes the sequence of TnaC and why many proteins that have sequences similar, but not identical, to that of TnaC do not shut down the ribosome. For more details, see our ribosome website.
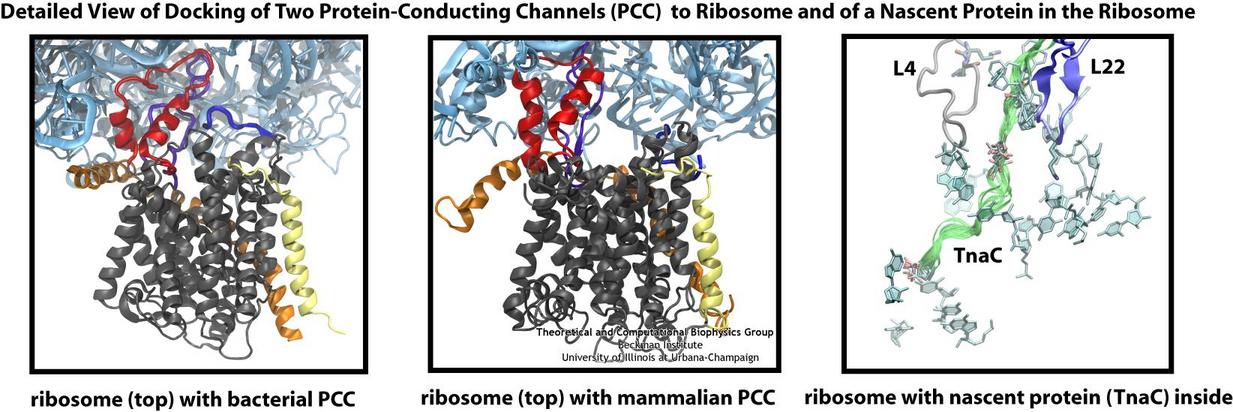
Living cells contain millions of proteins composed of sequences of different amino acids that typically fold spontaneously into well-defined three-dimensional conformations and then carry out their role as molecular machines serving manifold functions in the cells (see the protein folding highlight). The synthesis of the proteins is carried out by the ribosome, one of the largest molecular machines present in all cells, which reads the cell's genetic information for the purpose. Three researchers were recently awarded the 2009 Nobel Prize in Chemistry for the determination of the ribosome's structure. The physical mechanism of the ribosome, the cell's protein factory, is still largely unknown. Just as in any factory, there are multiple directions and controls on the protein assembly line. Sometimes the protein products need to be redirected to different parts of the cell, and other times assembly needs to be halted altogether. Now, significant new insights into both of these aspects of protein assembly have been made by combining electron microscopy with molecular dynamics simulations using the recently developed molecular dynamics flexible fitting method (MDFF, see the June 2008 highlight). This combination allowed researchers to visualize the complex between the ribosome and a protein-conducting channel that directs proteins into and across membranes for both a mammalian system (reported here) and a bacterial system (reported here). Amazingly, despite the evolutionary distance between mammals and bacteria, both complexes are remarkably similar. Simulations of the bacterial ribosome-channel complex, among the largest ever performed, further revealed the steps in the direction process. In a third study (reported here), researchers determined how TnaC, as a protein newly synthesized by the ribosome, can stall the ribosome from within during its own assembly, which then controls the expression of related genes. More information on these unique protein assembly controls can be found on our ribosome and our protein-conducting channel websites.
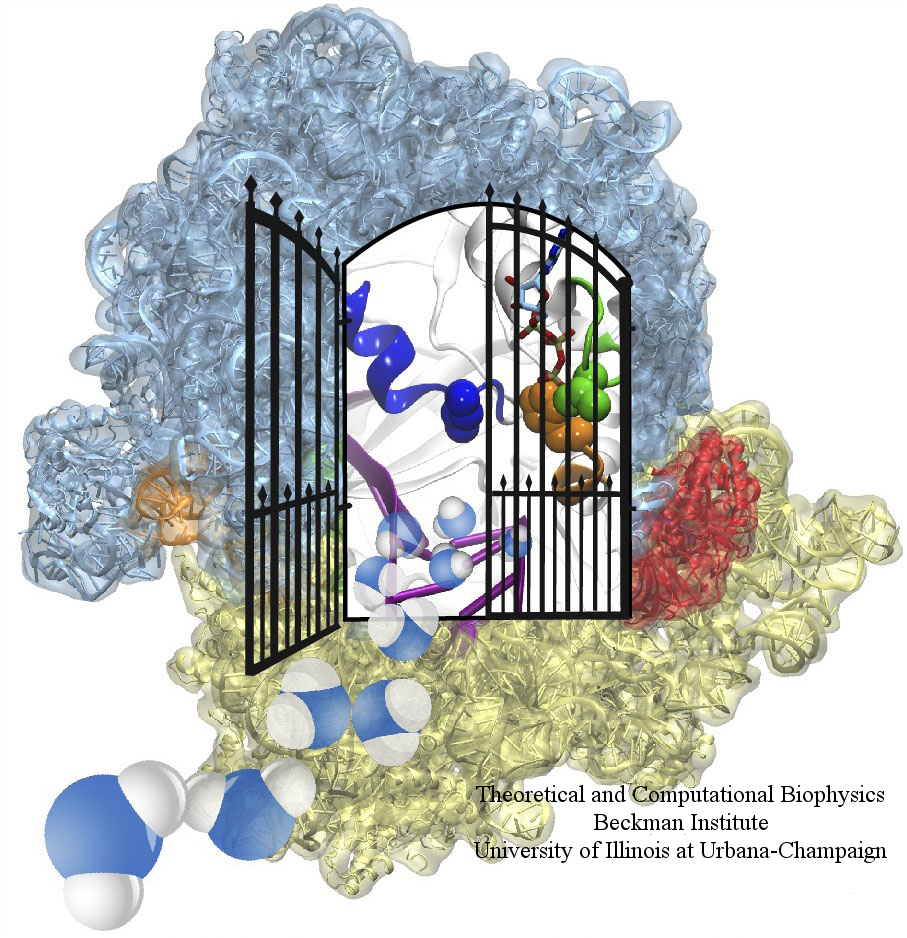
image size: 604.1KB
The ribosome is one of the largest molecular machines present in hundreds to thousands of copies in every cell, in charge of synthesizing every protein in the cell faithfully from genetic instruction. For this purpose the ribosome "reads" the sequence of bases on so-called messenger RNA, three bases at a time and depending on the base triple, the codon, elongates a nascent protein by one of 20 possible amino acids, avoiding to an impressive degree adding a wrong amino acid. So far one knew that the reading is done by transfer RNA molecules that have "foots" which match the possible codons and a "head" that brings along the associated amino acid. Each amino acid has its transfer RNA, the transfer RNAs checking if the next codon is "theirs," and if it is they add the proper amino acid to the nascent protein, elongating it. But how does the ribosome make the critical decision at the decoding center, namely if the transfer RNA "foot," the so-called anticodon, matches the codon? The answer is not known, but a key detail has now been discovered through a combination of electron microscopy and molecular dynamics simulation using NAMD, VMD, and a method called flexible fitting (MDFF, see the June 2008 highlight). It was known that a third molecular system is involved, called the elongation factor Tu (EF-Tu), which generates a key signal to the ribosome and transfer RNA through a chemical reaction. This reaction involves chemically attacking a substrate of EF-Tu, the molecule guanosine-triphosphate (GTP), with water, breaking a bond and turning GTP into guanosine-diphosphate (GDP). The puzzle was that EF-Tu is far away from the decoding center. The collaboration between experiment and simulation, reported here, revealed that "correct recognition" through anticodon-codon binding opens a gate in the EF-Tu that allows water access to the GTP inducing the signaling reaction. The finding promises to now establish how the decision at the decoding center is made and how an "open sesame" order is transmitted to EF-Tu. More on our ribosome website.
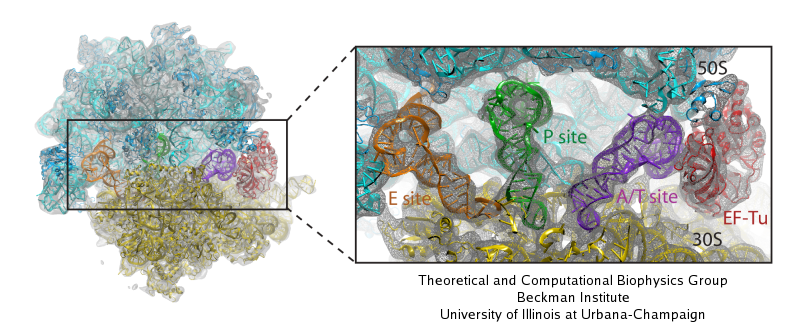
image size:
117.5KB
made with VMD
Living cells are brimming with activity, much of it due to their manifold molecular machines pulling cargo, importing and exporting molecules, digesting food molecules and transforming their energy, reading and copying genetic messages, or synthesizing proteins from these messages (the latter done by the ribosome). Static structures of the molecular machines have been resolved through crystallography: machines pressed into the confinement of crystals and frozen into inactivity reveal their atomic level geometry through this methodology. However, many machines, for example the ribosome, undergo large conformational transitions during their cyclic action, but active motions are hard to view in atomic detail. A way out is offered by electron microscopy which freeze-shocks machines into states characteristic for action cycle intermediates. Unfortunately, the method does not yield atomic resolution images, leaving the chemical detail needed for a comprehension of the mechanisms blurred. Computational methods can be used to bridge the resolution gap: atomic level structures of non-functional states of the machines captured in crystals are deformed to match electron microscopy images. Until recently, the method worked well only for very small machines. Now a team of electron microscopists and computational biologists using NAMD extended the method to common size machines and reported its successful application to the ribosome, providing astonishing detail about ribosome dynamics and function. For more details, see our MDFF website.


