Highlights of our Work
2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
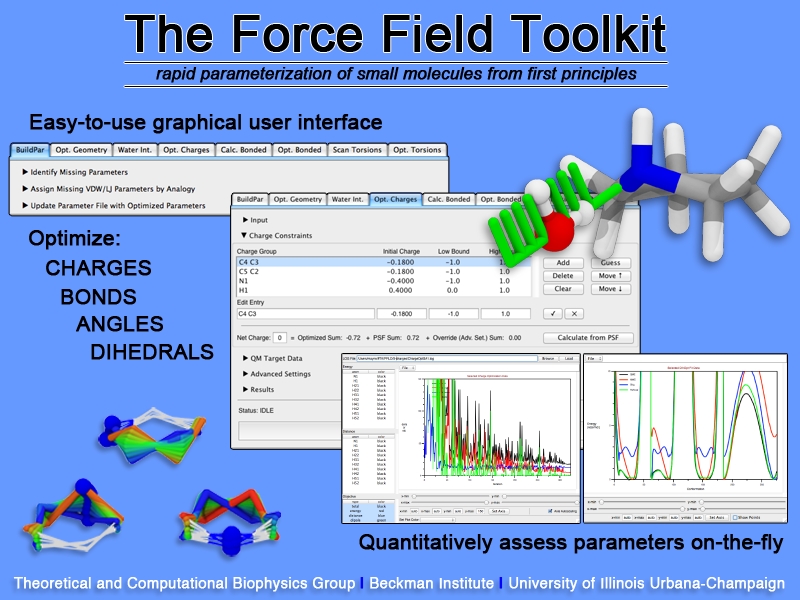
image size:
307.6KB
Structural biologists are increasingly turning to simulation methods to investigate the connections between molecular structure and biological function. Classical molecular dynamics (MD) simulations, such as those performed by the simulation software NAMD, rely on potential energy functions requiring parameters to describe atomic interactions within the molecular system. While these parameters are available for the most commonly simulated biopolymers (e.g., proteins, nucleic acids, carbohydrates), many small molecules and other chemical species lack adequate descriptions. The complexity of developing these parameters severly restricts the application of MD technologies across many fields, including most notably drug discovery. Recently, researchers have developed software, the Force Field Toolkit (ffTK), that greatly reduces these limitations by facilitating the development of parameters directly from first principles. ffTK, distributed as a plugin for the molecular modeling softare VMD, addresses both theoretical and practical aspects of parameterization by automating tedious and error-prone steps, performing multidimensional optimizations, and providing quantitative assessment of parameter performance--all from within an easy-to-use graphical user interface. Additional information on ffTK, including documentation and screencast tutorials, can be found here.


