Highlights of our Work
2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
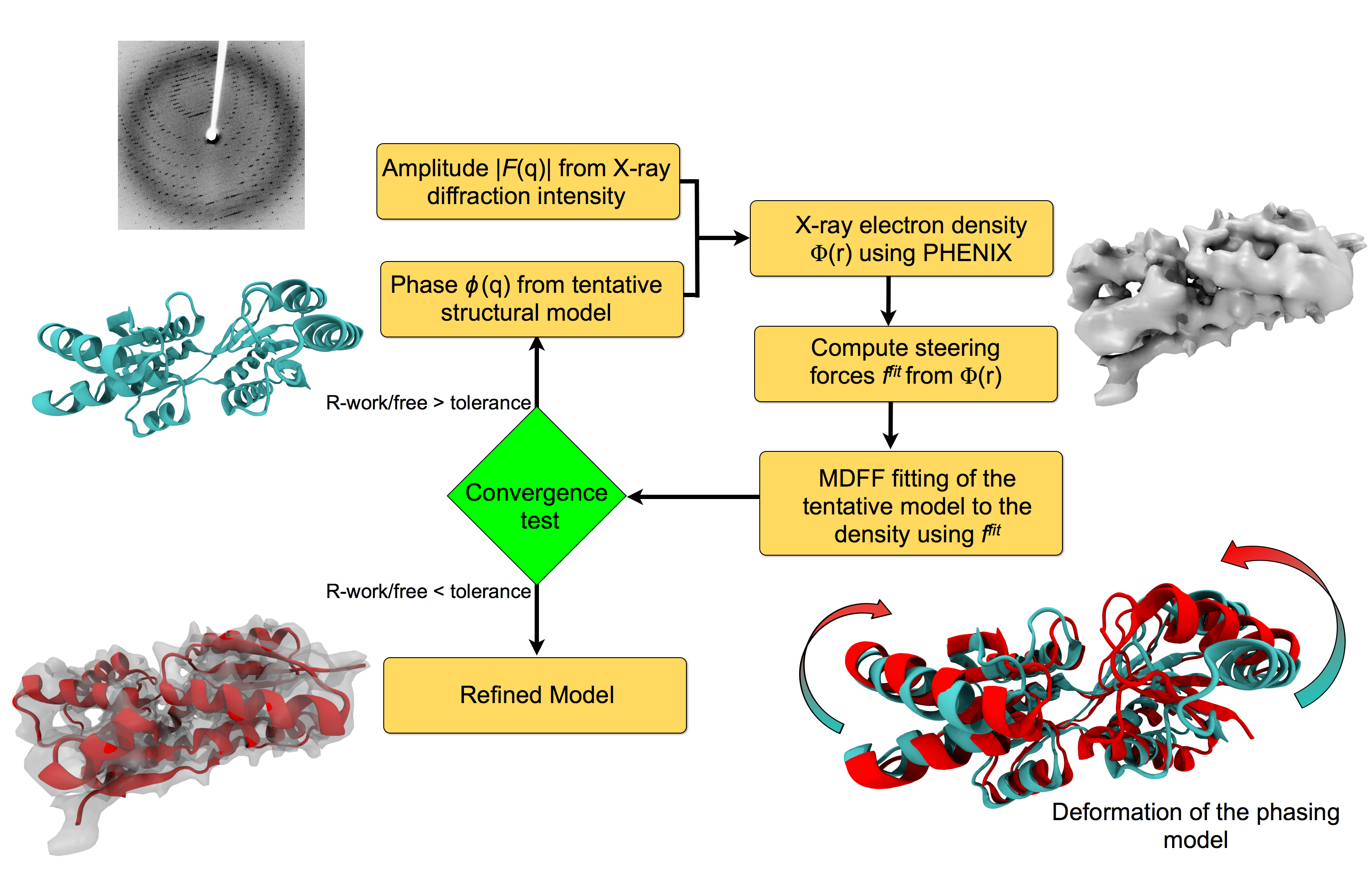
image size:
151.9KB
made with VMD
For many, the word 'X-ray' conjures up the images of white bones on black
backgrounds hanging on the wall of a doctor's office.
However, X-rays have played another important role for the past 100 years
through their use in the determination of chemical structures
at atomic level detail, starting with the first ever structure of
table salt in 1924. Since then, the diffraction properties of X-rays, when
shone on a crystal, have been used to solve increasingly large
and complex structures including those of biological macromolecules found inside living cells.
X-ray crystallography has become the most versatile and dominant technique for
determining atomic structures of biomolecules, but despite its strengths, X-ray crystallography struggles
in the case of large or flexible structures as well as in the case of membrane proteins, either of which diffract only at low resolutions.
Because solving structures from low-resolution
data is a difficult, time-consuming process, such data sets are often discarded.
To face the challenges posed by low-resolution,
new methods, such as xMDFF (Molecular Dynamics Flexible Fitting
for X-ray Crystallography) described
here,
are being developed.
xMDFF extends
the popular MDFF software originally created for determining atomic-resolution structures from
cryo-electron microscopy density maps (see the previous highlights
Seeing Molecular Machines in Action,
Open Sesame,
Placing New Proteins, and
Elusive HIV-1 Capsid).
xMDFF provides a relatively easy solution to the difficult process of refining structures from
low-resolution data. The method has been
successfully applied to experimental data as described in a
recent article
where xMDFF refinement is explained in detail and its use is demonstrated.
Together with electrophysiology experiments, xMDFF was also used to validate
the first all-atom structure of the voltage sensing protein Ci-VSP, as also
recently reported.
More on our
MDFF website.


