Research Topics - Steered/Interactive Molecular Dynamics
Knowledge of the mechanism of association, dissociation and unfolding of macromolecules is important for many biological processes. Among the examples are the binding and dissociation of substrates of enzyme reactions, the recognition of ligands by their receptors or the elastic resopnse of mechanical proteins. In order to study such processes external forces can be applied reducing energy barriers and therefore increasing the probability of unlikely events on the time scale of molecular dynamics. This approach has the advantage that it corresponds closely to micromanipulation through atomic force microscopy or optical tweezers. The external force techniques can be applied to study many processes, including dissociation of avidin-biotin complex, dissociation of retinal from bacteriorhodopsin, stretching of titin, etc. The molecular dynamics program NAMD, developed in the group, is capable of performing several different kinds of SMD, including rotation or translation of one or more atoms. The group's molecular graphics program VMD provides a powerful means of visualizing these simulations, and through the Interactive Molecular Dynamics (IMD) interface can even allow SMD simulations to be performed in real time.
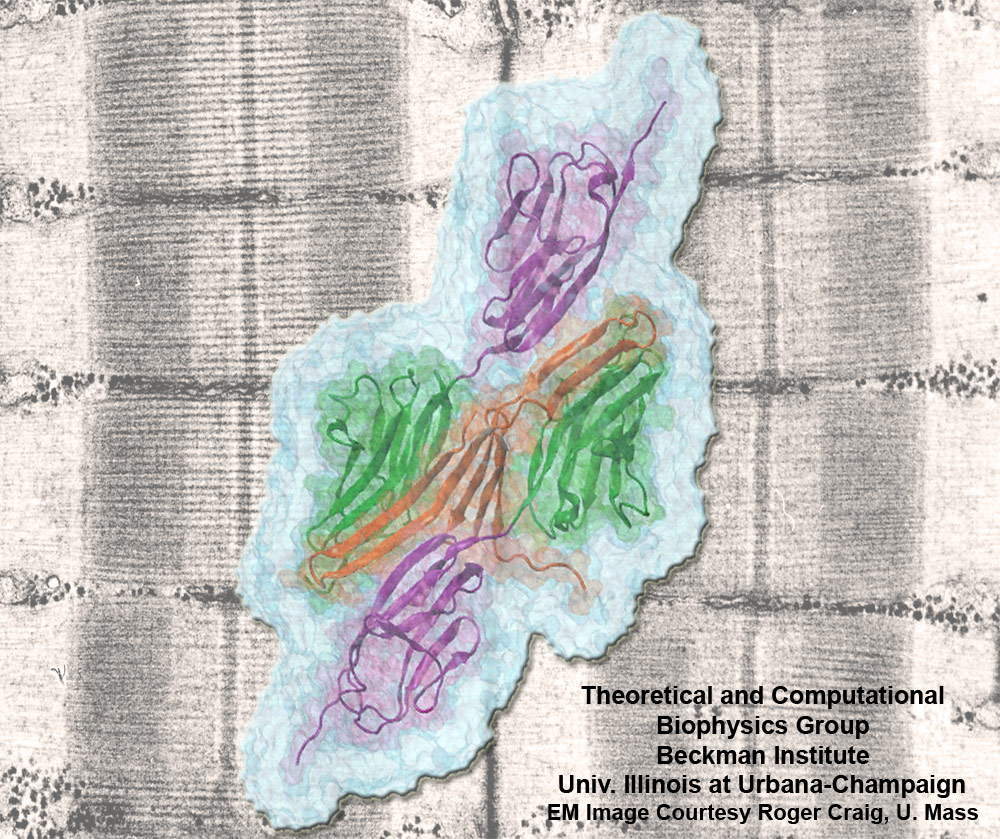
image size:
301.5KB
made with VMD
Muscle fibers are not rigid structures, but rather, they can both contract and extend in response to physiological demand. As a result, muscle sarcomeres must have a protective mechanism to prevent tearing and damage from overstretching. The giant protein titin fulfills this role by acting as a molecular rubber band, providing a passive resistance force during extension to restore the muscle fiber to its resting length. Conceivably, this rubber band must be anchored to a rigid structure in order to function. Biochemical investigations have speculated that the protein telethonin, located at the sarcomeric Z-disc, may serve this purpose. Genetic diseases related to defects in telethonin have been correlated with dilated cardiomyopathy and a form of muscular dystrophy. To date there have been no studies to determine how strongly bound titin is to telethonin. To explore this issue, we performed molecular dynamics simulations in order to test the strength of the newly resolved titin Z1Z2-telethonin complex. Our results, which have recently been reported (paper), reveal that the force required to dissociate titin from telethonin is significantly higher than that required to unfold isolated titin Ig-domains. This suggests strongly that telethonin is in fact an essential component of the Z-disc titin anchor. In addition, we find that telethonin anchors not just one, but two separate titin molecules, serving as a sort of molecular glue joining both titin molecules together through β-strand crosslinking (a structural motif also seen in fibril pathologies such as Alzheimer's, Parkinson, and Huntington's disease). Thus our simulations reveal also a fundamental architectural element of living cells, namely how cells glues their components together yielding strong mechanical connections. For more information on teletonin and the implications of our findings, see the following webpage here.
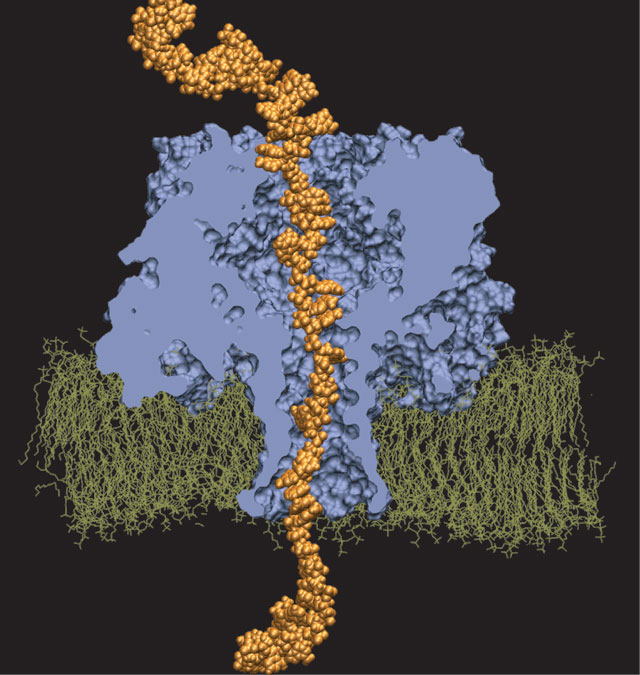
Interactive Molecular Dynamics allows us to pull sugar molecules by hand through a simulation of the glycerol channel GlpF. As we push the virtual molecules around, we feel them in our hands as if they were real. We use this technique to explore features of the channel and gain new insights into the way it functions.
image size:
301.5KB
made with VMD
Binding of the hormone to the retinoic acid receptor induces conformational changes that control and influence gene expression. In order to understand the functional role of the hormone one must understand the binding mechanism by which the hormone induces conformational changes. We studied the forced unbinding of the retinoic acid hormone from its receptor by applying an external force on the hormone.
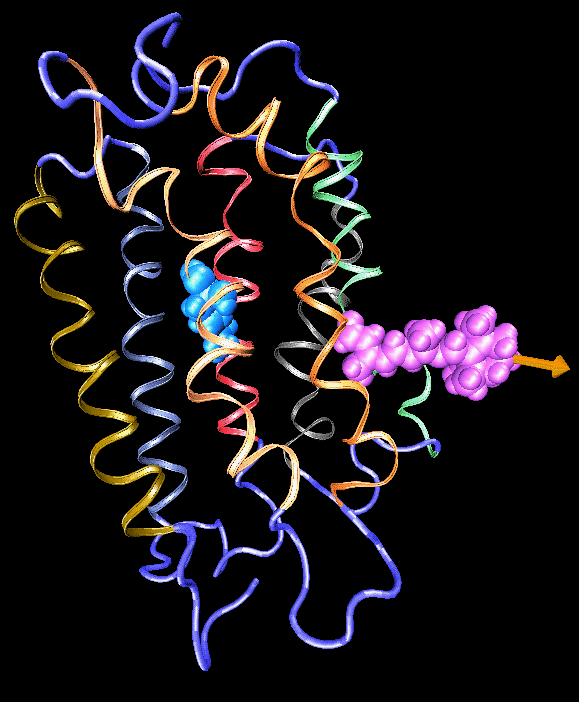
image size:
301.5KB
made with VMD
Formation of bacteriorhodopsin (bR) from the apoprotein and retinal has been studied experimentally, but the actual pathway, including the site of retinal entry, is little understood. Molecular dynamics simulations provide a surprisingly clear prediction.
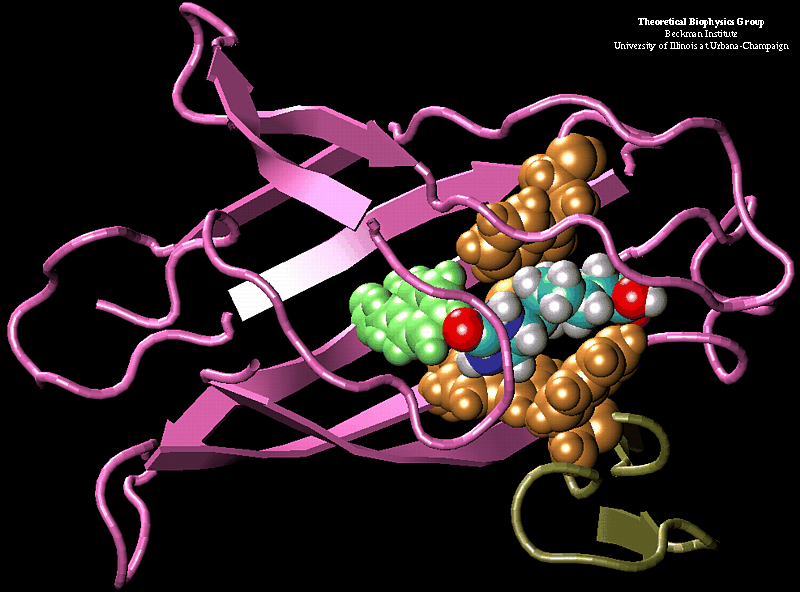
image size:
120.4KB
made with VMD
Molecular dynamics simulations induce, over periods of 40 ps to 500 ps, the unbinding of biotin from avidin by means of external harmonic forces with force constants close to those of AFM cantilevers. The applied forces are sufficiently large to reduce the overall binding energy enough to yield unbinding within the measurement time.
image size:
194.1KB
made with VMD
Bacillus anthracis, the cause of anthrax, is one of the most lethal bacteria. In addition to its ability to infect animal and human cells, the bacterium attacks also the cells of the host's immune system, the so-called macrophages. For this purpose the anthrax bacterium releases three types of proteins, or toxins, into the blood stream of the host: protective antigen, lethal factor, and edema factor, referred to as PA, LF, and EF, respectively. LF and EF team up with PA, which transports them into a host macrophage cell. Once inside the cell, LF converts ATP to cyclic AMP, while EF disables MAPKKs, a family of signaling proteins. These attacks disrupt various cellular signaling pathways of macrophages and some other cells, essentially shutting down the host's immune system and often leading to death of the host. To invade macrophages, the toxins take an intricate entry route that involves binding to a cell receptor, capillary morphogenesis protein 2 (CMG2), inducing the cell to internalize the toxins in a bubble like membrane (endosome), the bubble wall being then punctured by seven PAs forming a channel upon a chemical (acidifying) trigger from the host; the channel permits then their lethal cargo, LFs and EFs, to slip into the cell. How exactly the PAs punctured the endosome wall remained a mystery. In a recent report the entry route has been resolved now in greater detail through molecular dynamics simulations using NAMD. The report reveals how acidic conditions in the endosome trigger conformational changes of the PA complex necessary for pore formation, and provides structural insights into the role of unusual interactions between the PAs and its receptor CMG2. Visit also our anthrax toxin webpage.
image size:
215.8KB
made with VMD
image size:
299.9KB
made with VMD
image size: 468.6KB
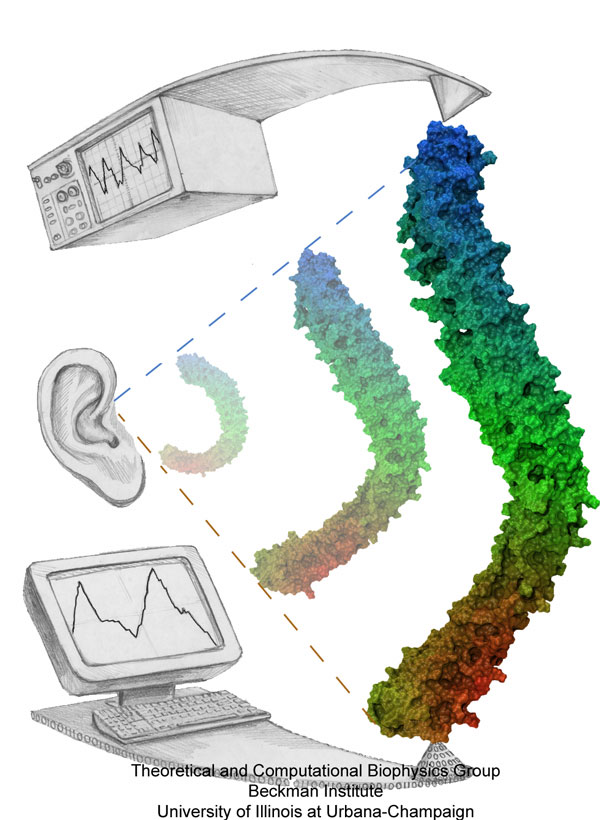
image size:
91.7KB
movie (
789.9KB
)
Mechanical forces are everywhere in human life. Strong forces power machines and cars, our body's forces let us labor and move, soft forces are sensed through touch, even softer ones through hearing. Forces are also ubiquitous in the living cell, driving its molecular machines and motors as well as signaling ongoing action in its surroundings. Man made, force bearing machines rely on extremely strong materials not found in the cell. How can the cell bear substantial forces? Also, how do cells sense extremely weak forces as in hearing, surpassing most microphones? Single molecule measurements begin to answer these questions offering information on biomolecules' mechanical responses and action. However, the information offered by these measurements is not enough to relate the biomolecular function to the biomolecular architecture. Biomolecules in cells can move in amazing ways, but we did not know why. As a review in Science demonstrates, computational modeling comes to the rescue. It can simulate the measurements and, in doing so, can reveal the physical mechanisms underlying cellular mechanics at the atomic level. In as far as observed data are available, the simulations show impressive agreement with actual measurements. While initially only following experiments or, at best, guiding experiments, modeling has advanced now further and through simulated measurements discovered on its own entirely novel mechanical properties that were later verified by experimental measurements. Experimentalists reacted to the new competition and began to do simulations themselves.
In a remote New Mexico dessert, Klaus Schulten met Amit Meller, who told him a story of two DNA hairpins. The first one, when threaded through the transmembrane pore of alpha-hemolysin blocks the ionic current to 12 pA and escapes from the pore about three times slower than the other one that blocks the current to only 9 pA. The amazing fact about these experiments was that the two DNA hairpins were identical in sequence. The only difference was in the global orientation of the single stranded part of the hairpin in the pore of alpha-hemolysin. Astonished by the outcome of their experiments, Amit Meller and Jérôme Mathé sought an explanation from the modelers (Klaus Schulten and Aleksei Aksimentiev). Cautious as the experimentalists are, they did not tell the modelers which of the DNA orientations produces the larger current blockade and which one escapes faster from the pore. We describe our quest for the solution of the puzzle.


