VMD Spotlights
We are continually adding new features to VMD (and expanding the
spotlight to highlight more of the existing features of VMD)
so come back and visit often.
|
Members of the Theoretical and Computational Biophysics Group
were part of a talented multi-institutional interdisciplinary team awarded
at Supercomputing 2020 with the internationally recognized
ACM Gordon Bell Special Prize for COVID-19 Research,
described as the "Nobel Prize for high performance computing."
The winning team developed a new AI-driven simulation workflow to
drive 8.5M-atom simulations of the SARS CoV-2 spike protein,
its interaction with the human receptor ACE2,
and 305M-atom simulations of the full SARS CoV-2 virion, using
NAMD on
Summit, the most powerful supercomputer in the United States,
operated by the Oak Ridge National Laboratory.
The NAMD simulations collectively performed over one ZettaFLOP
of calculation.
The AI-driven workflow and weighted ensemble methods used by the
team increased effective performance by orders of magnitude.
VMD was used to
build complete structures, prepare the simulations,
analyze the results, and create all of the team's visualizations
due to its unique capacity for working with
extremely large biomolecular systems and diverse structure data.
Due to decades of prior investment in the development of parallel
molecular dynamics, ongoing technology collaborations with
hardware vendors and national laboratories deploying top tier systems,
and our lab's prior work to enable large all-atom simulations of
viruses and other large systems of up to one-billion atoms, NAMD and VMD
were well-prepared for the challenges posed by the project
and its highly compressed research timeline.
The team set several new high-water marks for the full virion simulation,
molecular dynamics strong scaling performance, the
scale of weighed ensemble accelerated sampling, and the
use of AI-driven simulation for multiscale modeling.
This marks the second Gordon Bell Prize for NAMD, which was
also a winner in 2002.
Read more about the Gordon Bell Special Prize for COVID-19 Research here.
|
|
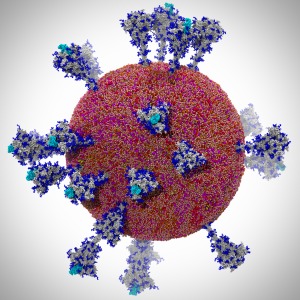
Image courtesy L. Casalino.
Made with VMD
|
|
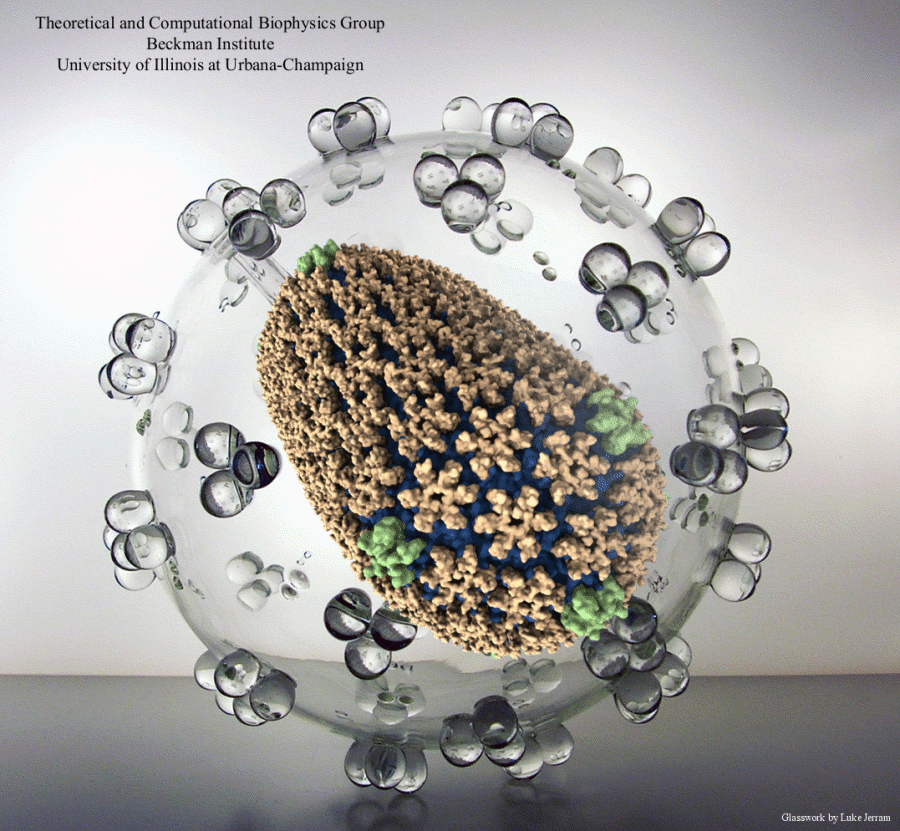
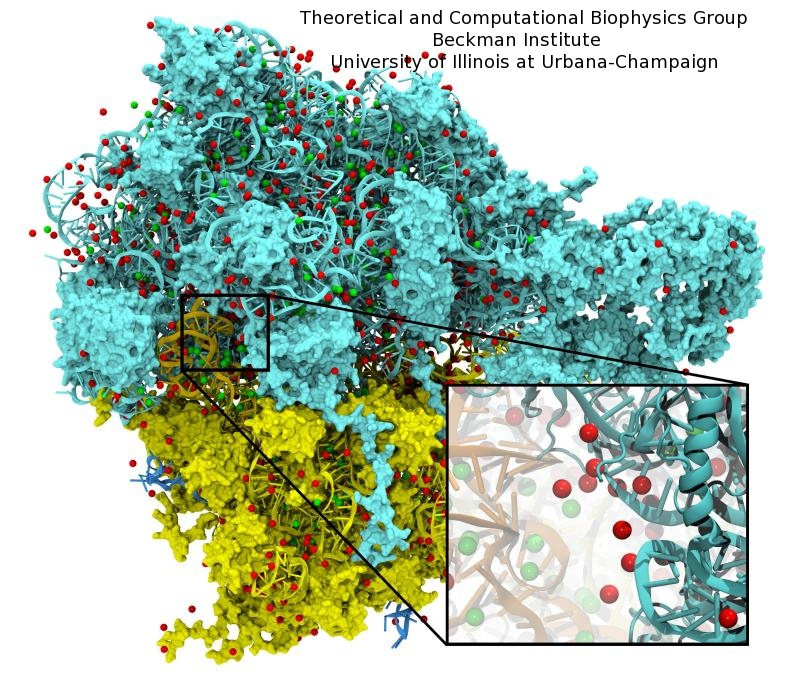
Disease due to a viral or bacterial infection continues to pose a major health concern, particularly today in the midst of the COVID-19 pandemic. Fighting such health threats requires the development of new antiviral and antibiotic drugs, a process that is increasingly guided by molecular modeling and computer simulation. The recently released NAMD 2.14 is now especially well suited to handle very large molecular systems for cell-scale simulations and to take advantage of massively parallel, GPU-accelerated architectures like the Summit supercomputer at Oak Ridge National Laboratory, currently ranked as the fastest in the world. Recent Summit simulations of a one-billion-atom protocell system, assembled to emulate a minimal but realistic cellular envelope and shown here in a cross-sectional view, push the limits of constructing and modeling atomic-resolution biological structures in VMD, paving the way for improved methodologies and tools that will enable the simulation of large viral and bacterial complexes.
|
|

|
|
Sunlight powers life on Earth. This basic fact has been known since ancient times and retold many times in many cultures. Today, scientists understand the means through which light powers life at atomistic detail, as a chain of processes climbing scales from electronic interactions to cooperation between proteins to cell-scale integration of energy conversion. These processes have been illustrated in a recent video
that was awarded the
BEST SCIENTIFIC VISUALIZATION OF 2019
at the SC19 conference, Scientific Visualization & Data Analytics Showcase.
The video is a joint production with the team of
Donna Cox
at
NCSA
and is based on a decade long collaborative effort with the experimental group of
Neil Hunter
and a lifetime research interest for
Klaus Schulten.
The underlying scientific investigation illustrated in the video
was presented in
18 manuscripts over the past decade,
from
atomic scale structural modeling
to
organelle-scale
and
cell-scale
integration of function.
These modeling efforts also led to
a molecular dynamics simulation of the chromatophore
using NAMD,
recently published in
Cell.
The video segment,
produced with VMD,
that won as the Best Scientific Visualization is an excerpt from
the fulldome movie
'Birth of Planet Earth' released to planetariums worldwide by Spitz Creative Media.
The oldest story of humanity — light powering life — coming soon to a theater near you.
|
|

image size: 621K
made with VMD
|
|
Modern molecular simulation, visualization, and modeling techniques often require high-performance computing hardware
to obtain the best efficiency. However, access to such hardware can be a barrier for some researchers.
As an alternative, cloud computing provides a cost-effective and practical solution for many molecular modeling tasks and for small and moderate size molecular dynamics simulations. The cloud computing model
provides researchers with access to powerful computational equipment
that would otherwise be too costly to procure, maintain,
and administer on their own. Additionally, cloud platforms can easily bundle different software packages
used in a modeling workflow to guaruntee their availablity and
interoperability on a standardized system.
We have adapted our molecular modeling applications NAMD, VMD, and associated tools to
operate within the Amazon Web Services (AWS) Elastic Compute Cloud (EC2) platform, enabling popular
research workflows to be run remotely by scientists all over the globe,
with no need for investment in local computing resources and
a reduced requirement for expertise in high performance computing technologies.
Recent advancements in GPU virtualization technology now make it possible to use cloud computing for
large-scale scientific computing, data analysis, and visualization tasks. The latest release of our
cloud software can be run on EC2's latest graphics hardware,
backed by DCV, to provide a smooth and seamless
graphical working environment. Instance types specially designed for parallel CPU or GPU workloads
provide users with access to the hardware they need to run even the most demanding features of NAMD and VMD,
all from their much less powerful personal computers and even laptops. These instances can be paid for on an on-demand, as-needed basis, and some
workflows, such as Molecular Dynamics Flexible Fitting, can be run for less than the cost of a cup of coffee.
Our cloud virtual machine image is availble in the Amazon Marketplace,
which provides users with
a very simple one-click launch using pre-configured choices
of instance types that have been tested with our software.
To learn more,
see our
cloud research page.
|
|

|
|
In 2017, the Royal Swedish Academy of Sciences awarded the Nobel Prize in Chemistry to
Jacques Dubochet,
Joachim Frank, and
Richard Henderson
"for developing cryo-electron microscopy for the high-resolution structure determination of biomolecules in solution".
We are pleased to celebrate this great triumph for structural biology along with the well-deserved recognition of the Center's long-time collaborator and friend, Joachim Frank.
Our center has a long tradition in developing computational methods that enable scientists to build atomistic models of biomolecules.
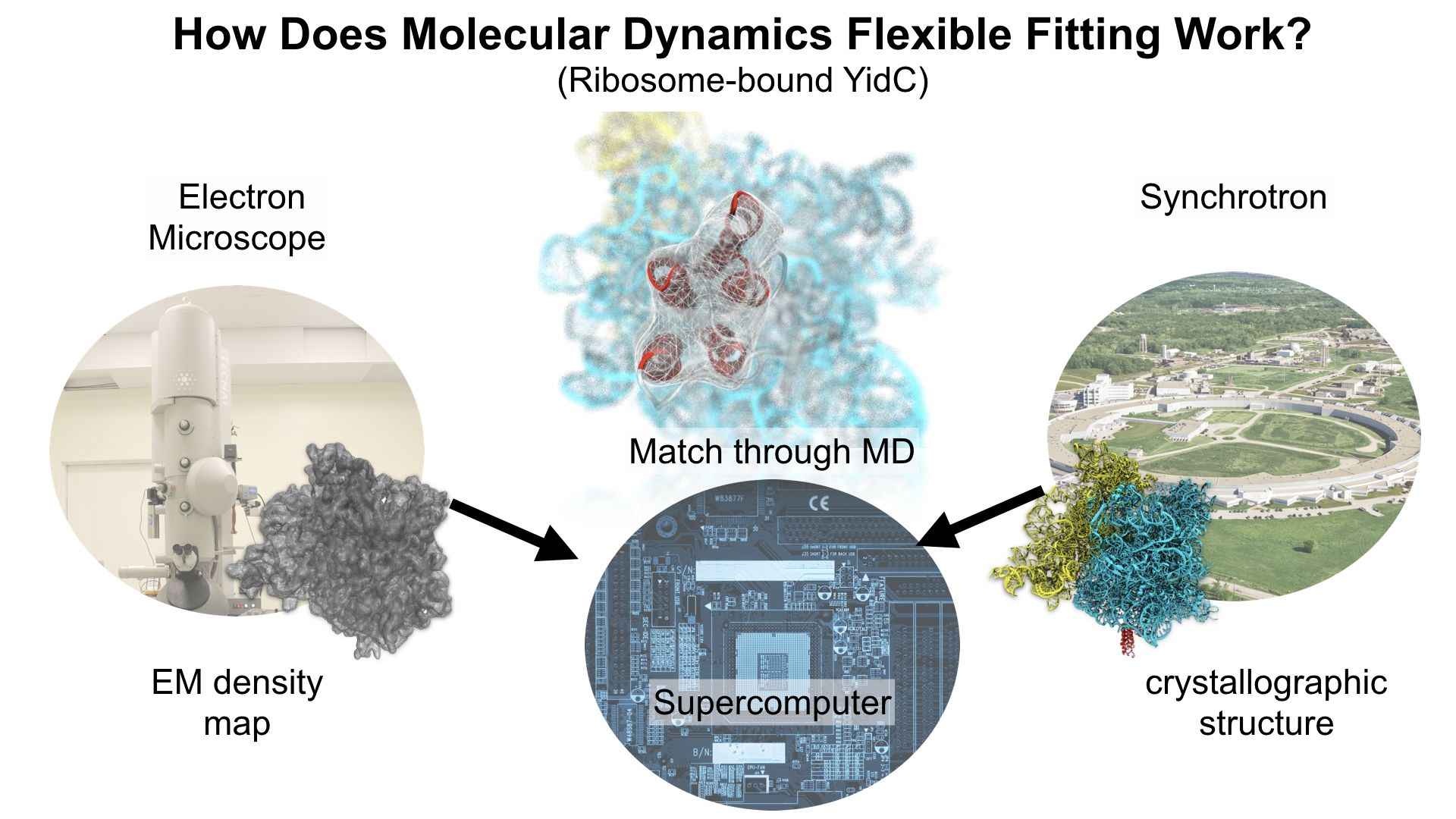
Molecular Dynamics Flexible Fitting (MDFF),
a method developed in close collaboration with Joachim Frank and his group,
reconciles high resolution data from X-ray crystallography and functional information from cryo-electron microscopy (cryo-EM).
MDFF utilizes molecular dynamics to "naturally" fit each atom into a cryo-EM map.
In less than a decade since its development, MDFF has proved instrumental in studying biomolecular systems.
A selected list of publications employing MDFF both by our group and others can be found
here.
|
|
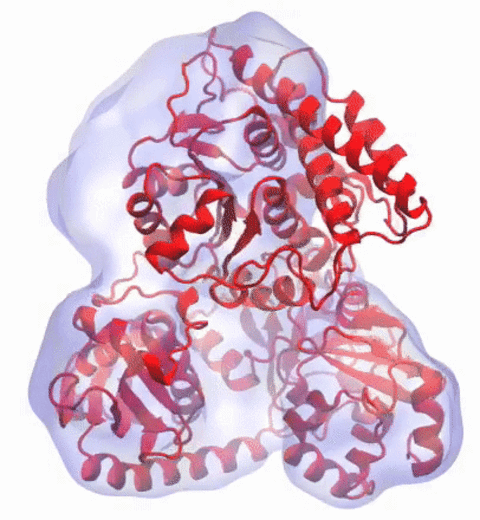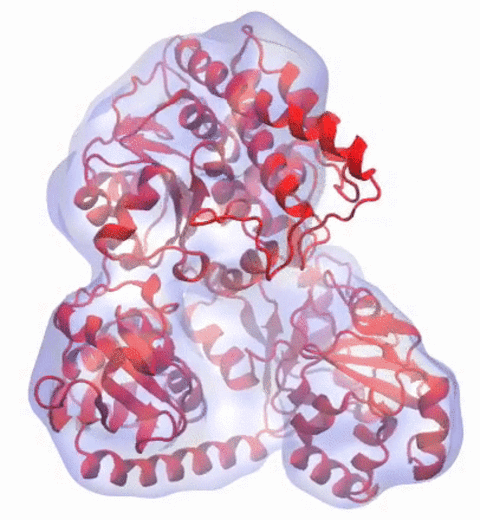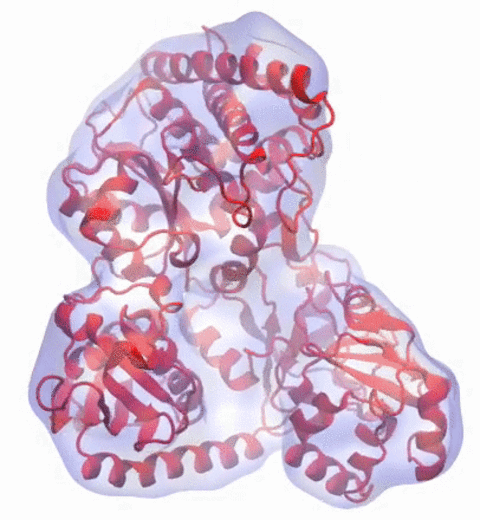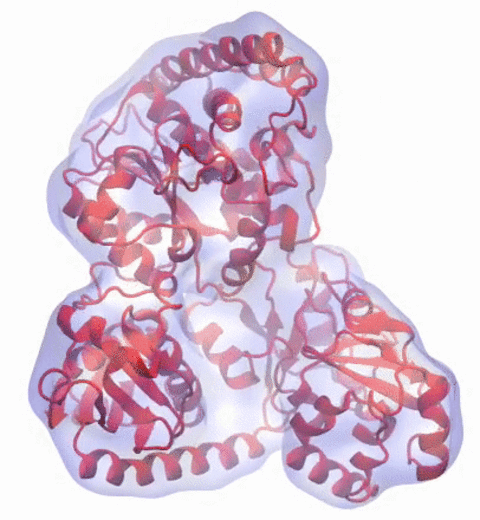
image size:
6.8M
made with VMD
|
|
On April 20th, 2017, the Journal of Physical Chemistry published a Memorial Issue in honor of Klaus Schulten gathering more than 60 research articles. Klaus was an undisputed leader in theoretical and computational biophysics, recognized by his peers for his immense contribution to the field, and having devoted his entire career to establish the mechanisms that underlie cellular processes using the laws of physics. Originally planned to be a Festschrift to celebrate Klaus's achievements on his 70th birthday, the Memorial Issue initiative immediately triggered a unanimous positive response from friends, academic colleagues and longtime collaborators across the world. The many contributions assembled in the Memorial Issue of the Journal of Physical Chemistry lie at the confluence of theory and experiment, and cover a broad gamut of topics that were dear to Klaus, ranging from photosynthesis to molecular machines and membrane proteins. We gratefully acknowledge the many authors of the Memorial Issue, who enthusiastically accepted to pay one last homage to Klaus through contributions of very high scientific quality.
|
|

image size:
213K
|
|
The 2.12 release of
the molecular dynamics program NAMD
provides major enhancements in performance, flexibility, and accuracy,
complementing the greatly enhanced usability provided by the
QwikMD GUI released in
VMD 1.9.3.
NVIDIA GPU-accelerated simulations with NAMD 2.12 are up to three times as
fast as 2.11, particularly for implicit solvent simulations and
single-node simulations of smaller systems.
NAMD 2.12 is also optimized for the new Intel Xeon Phi KNL processors found in
Argonne Theta,
NERSC Cori,
and
TACC Stampede 2.
NAMD 2.12 builds on the asynchronous multi-copy scripting capabilities introduced in
NAMD 2.11
with the ability to modify and reload the molecular structure,
enabling development of grand canonical and constant pH ensemble methods,
as well as an optional Python interface for advanced on-the-fly analysis.
Finally, NAMD 2.12 provides a complete, no-recompilation-needed
interface for hybrid QM/MM
with both the semi-empirical code MOPAC and the ab initio/DFT code ORCA.
More on new features in the 2.12 release of NAMD can be found
here.
NAMD is available free-of-charge as source code, precompiled binaries,
pre-installed at supercomputer centers, and now jointly with VMD as
one-click interactive molecular modeling
on the Amazon cloud.
|
|
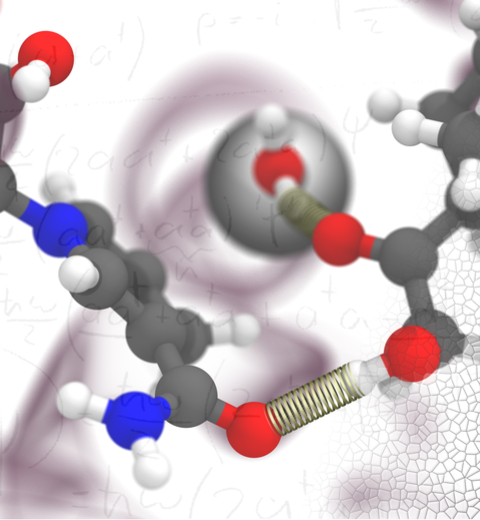
image size:
508K
made with VMD
|
|
The latest release of VMD brings many
advances that help researchers prepare, analyze, and visualize
molecular simulations.
The new
QwikMD plugin
streamlines key simulation preparation and analysis tasks, and guides users
in the creation of reusable simulation workflows and protocols.
VMD now includes several advanced features for parallel analysis
and visualization of cellular-scale simulations, as
reported here,
and here.
VMD 1.9.3 strengthens collaboration between experimental and computational
biologists by supporting a broader range of experimental density map
image formats, such as those used in cryo-electron tomography.
Many updated plugins are included in VMD 1.9.3, including tools for
analysis of free energy perturbation simulations,
MDFF hybrid structure fitting,
ffTK force field parameterization,
and
normal mode analysis.
VMD 1.9.3 adds support for new hardware and operating system
platforms including
IBM OpenPOWER (ORNL Summit),
a variety of GPU-accelerated ARM SoCs,
the Amazon AWS EC2 cloud,
and most recently, the Intel Xeon Phi Knight's Landing many-core CPU (TACC Stampede 2, Argonne Theta).
The VMD 1.9.3 release adds stunning graphics produced using
interactive ray tracing using the latest multi-core CPUs and GPU accelerators,
enabling 360-degree panoramic movie rendering for VR headsets,
as
reported here,
and here.
Interactive ray tracing makes the task of getting a molecular
image "just right" much easier than ever before; it also enables
rendering of spectacular movies for communication of scientific results.
A VR movie rendering tutorial
assists users with the steps required in rendering and encoding
VR movies for upload to YouTube for display using VR headsets such
as Google Cardboard, Oculus Rift, and GearVR.
More details about VMD 1.9.3 features can be found
here.
|
|

image size:
2.2M
made with VMD
|
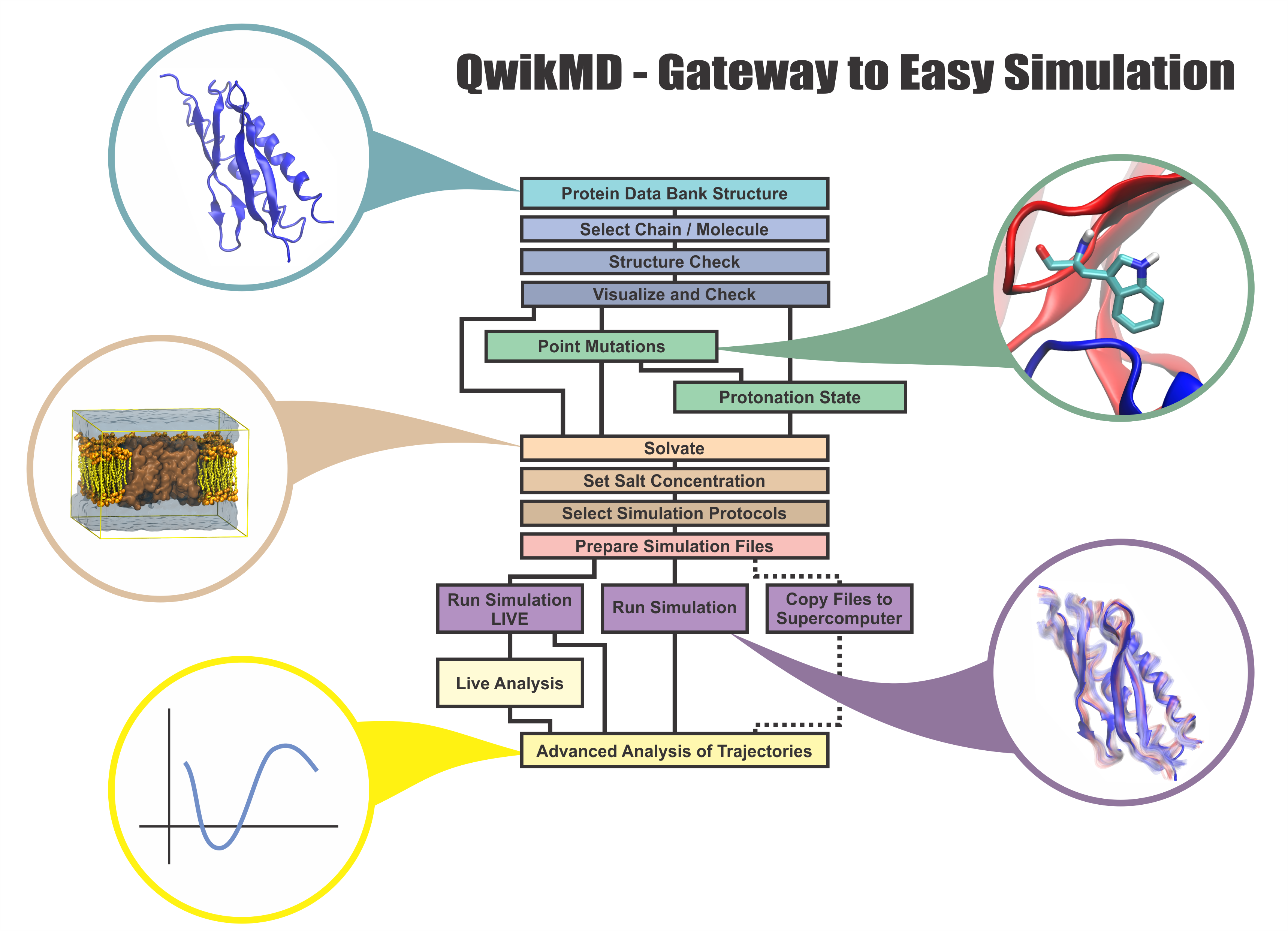
Everything that living things do can be understood in terms of jigglings and wigglings of atoms.
Richard Feynman's remark in the early 1960's summarizes what is today widely accepted, namely,
that molecular processes can be described by the dynamics of biological molecules, therefore connecting
protein dynamics to biological function. Molecular dynamics (MD) is by far the best tool to investigate
jigglings and wigglings of biological systems. Advances in both software and hardware
have spread the use of MD, however the steepness of the learning curve of the methodology of MD
remains high. To assist new users in overcoming the initial barrier to use MD software, and to help the more advanced users to
speed up tedious steps, we have developed the QwikMD software, as decribed in a recent paper.
By incorporating an easy-to-use point-and-click user interface that connects the widely used molecular graphics
program VMD
with the powerful MD program NAMD,
QwikMD allows its users to prepare both basic and advanced MD simulations in just a few minutes. At the same time,
QwikMD keeps track of every step performed during the preparation of the simulation, allowing easy reproducibility
and shareability of protocols. More information about QwikMD, as well as introductory tutorials are available on our
QwikMD webpage.
QwikMD is available in VMD 1.9.3 or later versions. |
|
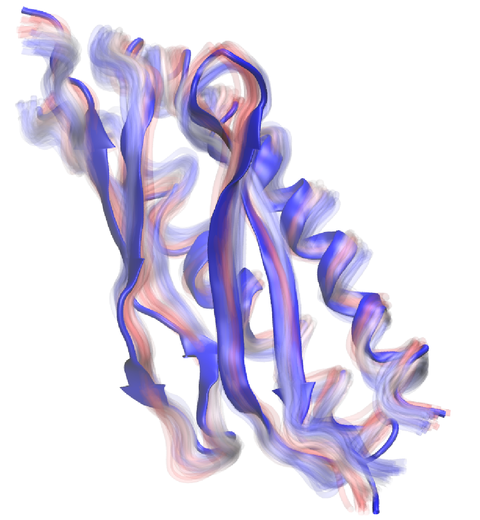
image size: 1.2M
made with VMD
|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow large-size and
long-timescale simulation trajectories to be loaded into physical memory
and accommodate large volumetric datasets.
The 64 million atom HIV capsid simulation
described in the May 30, 2013 issue of Nature is a first-class
example of what can be done with VMD on an appropriately equipped
graphics workstation. The HIV-1 model was prepared for simulation
using the structure building tools and scripting features of VMD were
later used for trajectory analysis. The all-atom structure of the
HIV-1 capsid shown on the Nature cover was rendered within VMD
using the internal
Tachyon
ray tracing engine, and was then composited with an artistic
representation of the viral envelope and the Nature cover text.
|
|

|
|
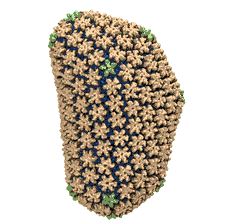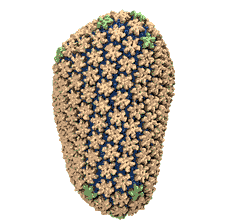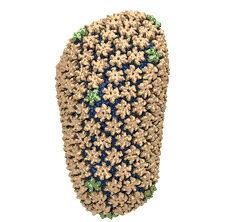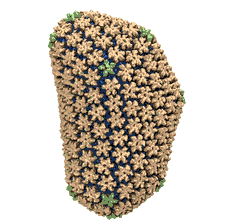
Human immunodeficiency virus type 1 (HIV-1) is the major cause of
AIDS, for which treatments need to be developed continuously as the
virus becomes quickly resistant to new drugs. When the virus infects
a human cell it releases into the cell its capsid, a closed, stable
container protecting the viral genetic material. However, interaction
with the cell triggers at some point an instability of the capsid,
leading to a well timed release of the genetic material that merges
then with the cell's genes and begins to control the cell. The dual
role of the capsid, to be functionally both stable and unstable, makes
it in principle an ideal target for antiviral drugs and, in fact,
treatments of other viral infections successfully target the
respective capsids. The size of the HIV-1 capsid (about 1,300
proteins), and its irregular shape had prevented so far the resolution
of a full capsid atomic-level structure. However, in a tour de force
effort, groups of experimental and computational scientists have now
resolved the capsid's chemical structure (deposited to the protein
data bank under the accession codes 3J3Q
and 3J3Y).
As reported
recently (see also journal cover), the
researchers combined NMR structure analysis, electron microscopy and
data-guided molecular dynamics
simulations utilizing VMD to prepare
and analyze simulations performed using NAMD on one of the most powerful computers
worldwide, Blue
Waters, to obtain and characterize the HIV-1 capsid. The
discovery can guide now the design of novel drugs for enhanced
antiviral therapy.
More information is available on our virus website, in video, and in a press release.
|
|
See also movie: 15.5MB
made with VMD
|
|
VMD is frequently used to make figures and illustrations that grace
the cover pages of textbooks and journals.
The cover image on the August 2008 issue of Science
by Klein and Shinoda demonstrates the use of VMD for visualization of large coarse-grain molecular dynamics trajectories.
The image illustrates a simulation of a vesicle interacting with a lipid bilayer, with
over 1 million coarse-grained interaction sites, equivalent to over 10 million atoms.
Read more about their work here.
|
|

|
|
Since its inception, VMD was designed to take advantage of
graphics processing units (GPUs) for interactive renderings of large
biomolecular complexes. Recently, GPUs have become programmable and
their massive parallel processing capabilities can now be utilized
for non-graphical computations. VMD makes uses of this type GPU-based
parallel computation to greatly accelerate calculation of electrostatic
potential fields, used for visualization and analysis,
and for modeling operations such as ion placement.
Ongoing VMD development efforts will expand the use of GPU acceleration
even further, for acceleration of volumetric processing,
structure and trajectory analysis, and compute-limited operations
performed within VMD and it's plugins.
More information on GPU acceleration of molecular modeling
applications is provided
here.
|
|

|
|
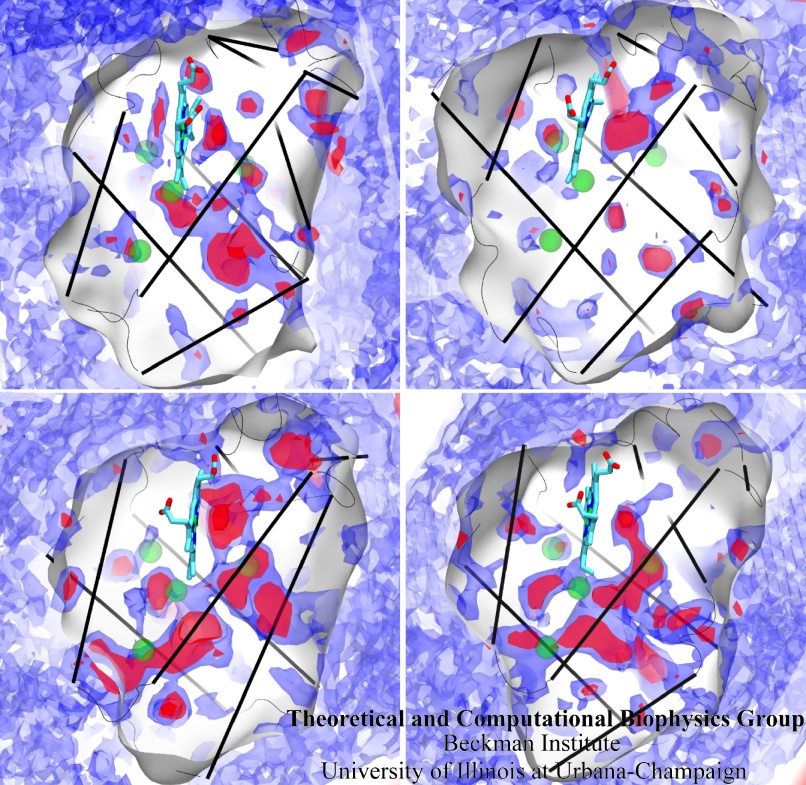
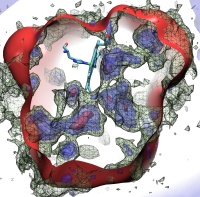
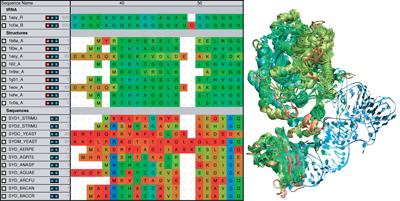
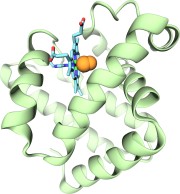
Globins are oxygen-storing proteins, vital to life. In our blood,
hemoglobins carry oxygen from our lungs to every cell in our body. In our
muscles, myoglobins keep reserves of oxygen to make sure it is available
when needed. In some plants, leghemoglobins capture oxygen molecules that
would otherwise be harmful to the production of ammonium necessary for the
plant's survival. All these globins possess an iron-containig "heme", that
grabs on to oxygen for a short time, and share the same protein
architecture, despite large variations in their sequences. Since the heme
group is buried inside a globin, scientists wondered how oxygen makes its
way inside the protein to reach it.
Exploring the motion and energetics of globins using the program NAMD researchers learned to gather data that
permitted them to visualize, utilizing the VMD software, all the pathways taken by oxygen
migrating inside whale myoglobin (see the Aug 2006 highlight and related
publication). However, when the researchers turned their attention to
the rest of the globin family to compute their oxygen pathways, they
found, on their computational spelunking trip,
something surprising. Given the conserved architecture of all globins,
they expected to see similar oxygen pathways throughout the globin family,
but they saw the opposite! Aside from a conserved pocket right at the
heme binding site, the distribution of oxygen pathways showed very little
similarity from one globin to the next. This result is described in a
recent report, which
shows that oxygen-pathways are not conserved by evolution, and that their
location is not determined by a protein's overall architecture, but rather
by its local amino acids. The researchers also learned which amino acids
are found more often than others lining oxygen pathways, recognizing that
bulky side groups are not hindering, but favoring oxygen passage. More
information can be found here.
|
|
|
|
Modern computers include a massively parallel graphics
processing unit (GPU) designed to perform geometry and lighting
calculations at blistering speeds.
State of the art GPUs can perform 0.5 teraFLOPS with their
hundred cores.
The tremendous computational power of GPUs
was untapped by scientific computations because it could only be
accessed with difficulty until now.
As
reported in the Journal of Computational Chemistry, recent
advances
allowing GPUs to be used for general purpose computing have
boosted the performance of a number of molecular modeling applications,
including
NAMD
simulations and
VMD
electrostatic potential calculations.
The accelerated versions of these
applications run five to one hundred times faster than on the best
CPU-based hardware, allowing a single desktop computer equipped
with a GPU to provide processing power equivalent
to an entire, large computing cluster.
More information on GPU acceleration of molecular modeling
applications is provided
here.
|
|
|
|
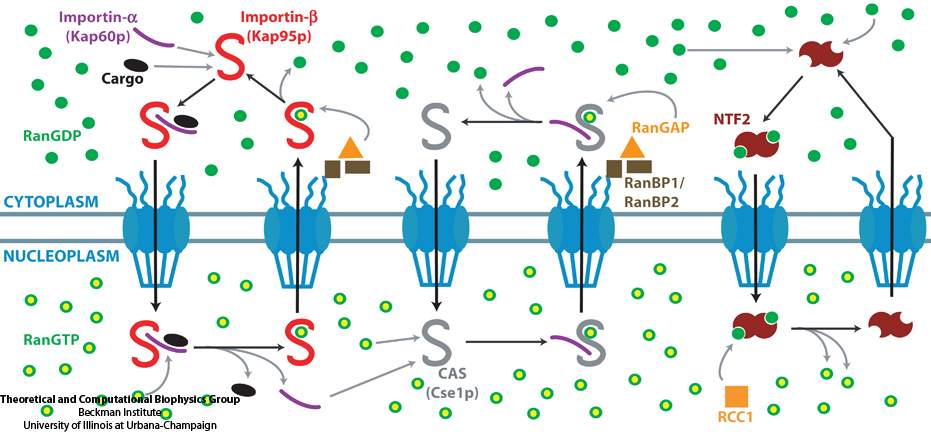
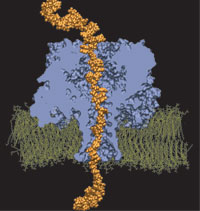
The cells of higher organisms store their genetic material, the genome, in the so-called nucleus where they organize transcription of DNA into messenger-RNA, the blueprint for proteins. The messenger-RNA leaves the cell to be decoded by ribosomes that synthesize the respective proteins. Transcription factors, also proteins, control in the nucleus which parts of the cells' genomes are transcribed. Naturally, the access to the nucleus as well as exit from it must be restricted to transcription factors and related biomolecules. This is achieved by the nuclear pores, wide channels lined with brushes of polymers. The polymers are disordered proteins and prevent passage for most cellular proteins, except for so-called transport factors which bus transcription factors, messenger RNA, and certain larger biomolecule into and out of the nucleus. How transport factors are permitted to pass the nuclear pores, despite many studies, has been largely unknown. Molecular dynamics simulations, based on relevant crystallographic structures, using NAMD provided a comprehensive picture on the passage mechanism as reported recently. The simulations, analyzed with VMD, revealed that transport factors are dotted rather regularly on their surface with spots that bind to the brushes of nuclear pore proteins. While any protein may accidentally exhibit such a binding spot or two, only transport factors offer a regular pattern of such spots on their surface that apparently is their passport permitting them movement into and out of the nucleus, i.e., helping them to glide through the pores' protein brushes. More on simulations of transport factors can be found here. |
|
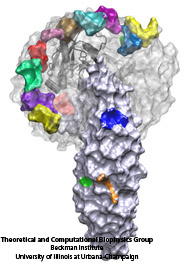
image size: 245K
movie: 4.6MB
made with VMD
|
|
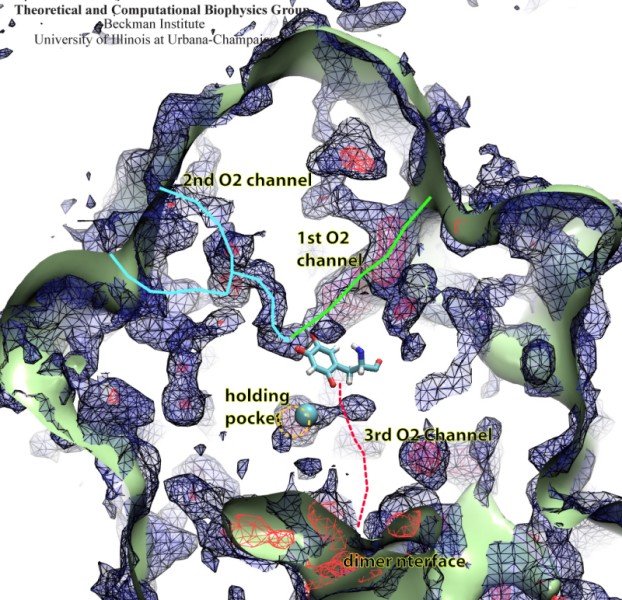
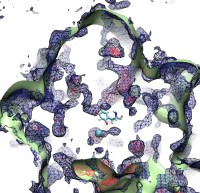
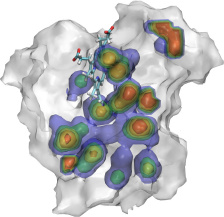
Because oxygen gas is very reactive, it is frequently employed by the cell
as a reagent by proteins called enzymes, which build the organic compounds
that the cell needs. One such enzyme belongs to the copper amine oxidase
family. These proteins transform amine-containing compounds into molecules
needed by the cell, by reacting the compounds with oxygen. Researchers
have long been interested in finding out how the various reagents reach
the buried copper active site before the final oxidation reaction can
occur. While copper amine oxidases exhibit an obvious channel for
capturing the amine compounds to be modified, it had been unclear until
now how oxygen molecules make their way through the enzyme. With the help
of computer simulations using NAMD,
researchers have identified in a recent publication, the
routes taken by oxygen inside various copper amine oxidases from different
species. In order to accomplish this, they analyzed simulations of the
motions of four copper amine oxidases, using the VMD analysis and visualization software, which
can predict the probability of finding oxygen molecules anywhere inside
the simulated proteins. This analysis found numerous oxygen conduction
routes inside each copper amine oxidase, i.e., oxygen can enter the
protein through many routes, as it would in a sponge. More information on
finding O2 migration pathways in proteins can be found here.
|
|
|
|
The popular Adobe PDF document format (PDF version 1.6) now supports
inclusion of 3-D geometry. The latest versions of Adobe Acrobat Reader
can now display, rotate, and animate 3-D models embedded within these
PDF documents. This capabilities provide a tremendous new opportunity
for creating tutorials, presentations, and electronic textbooks containing
3-D models of biomolecules that can be interactively manipulated by
the reader. For more information, see the
short tutorial on creating 3-D PDF files with VMD
|
|
|
|
The
Tachyon
parallel ray tracer included with VMD has been updated with
the ability to display molecules with ambient occlusion lighting.
This lighting technique mimicks some of the effects that occur under
conditions of omnidirectional diffuse illumination, e.g. outdoors on an
overcast day. This lighting used in concert with traditional point
source lights and directional lights to increase the perception of shape and
depth in images of molecular structures, and decreases the deleterious
effects of harsh shadows which otherwise occur with lighting based on
a small number of direct light sources. This lighting model is particularly
good at emulating the appearance of chalky materials, and can make
molecular graphics more understandable, not to mention aesthetically appealing.
A short
tutorial is available
on using these new lighting and shading features with VMD, along with a number
of representative example images.
.
|
|

|
"I would add that VMD (and similar tools, if they yet exist) will
become a primary platform upon which to study a wide range of
biological problems in the future. VMD adds a new dimension to the
biologist's thinking. Certain thoughts generated in interaction with
VMD images are inconceivable otherwise; the `language' essential to
generating these ideas would just not be there! In other words, VMD
is far from a simple `visualization tool' for a biologist, it is a
true thinking tool. Without it a whole class of biological
hypotheses would simply not exist."
-- Carl Woese
|
|
|
|
VMD supports user-defined material and shading properties that
can be used to render molecular graphics in a more illustrative
style. Future versions of VMD will expand on this capability
through increased use of programmable shading technology. This will
bring many molecular rendering features previously found only in batch mode
software renderers into the realm of interactive molecular visualization.
|
|
|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow huge simulation
trajectories to be loaded into physical memory and accomodate large
volumetric datasets.
This 1,000,000+ atom Satellite Tobacco Mosaic Virus simulation
is an example of the size simulation trajectory that one can analyze and
display with VMD on an appropriately equipped graphics workstation.
|
|

|
|
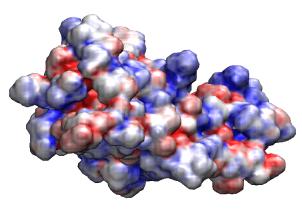
VMD includes a plugin for creating and running
APBS electrostatics calculations
and can
display the resulting ouput
including potential maps, solvent accessibility maps,
and other data produced by the APBS job. Recent versions of VMD also have
the ability to run APBS jobs on remote clusters or supercomputers for fast
turnaround of high resolution potential calculation needed for visualization
and analysis.
|
|
|
|
VMD can load and display volumetric data sets, including
electron density maps, electron orbitals, potential maps,
and various types of user-generated volumetric data.
The VMD plugin library
contains support for a large number of volumetric file formats.
Users can also import their own data file formats by writing
their own loader scripts using the "mol volume" command.
Volumetric data can be rendered using "VolumeSlice" or "Isosurface"
representations, each of which provides several geometric rendering
styles for viewing the data, varying isolevels, slice plane position,
etc.
|
|
|
|
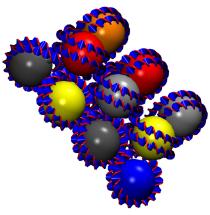
VMD includes a Virtual DNA Viewer
plugin which can be used to render images of chromatin using ideal DNA
interbasepair helical parameters (in proper r.h.s. ref. frame ordering).
This plugin is useful for investigating the folding of chromatin based on the
"two-angle" model where the total linker length and twist between nucleosomes
determines the overall geometry of chromatin. This is an example of the
type of visualizations that can be done with VMD using mathematical
models and equations rather than all-atom structure data.
|
|
|
|
VMD can be used to
display the results
of HOLE calculations. HOLE
calculates pore dimensions of the holes through molecular structures of
ion channels. This is one of many examples of how third party packages
can make use of the visualization capabilties of VMD.
|
|
|
|
Recent improvements to the data structures and algorithms used to
display and analyze molecular structures in VMD have improved
it's utility in
bionanotechnology,
and materials science where large structures, various types of non-biological
matter, and non-polymeric structural elements are often present.
|
|

|
|
Dr. Oliver Beckstein's VMD image was voted "highly commended" in the
2005 Visions of Science Photographic Awards.
The image illustrates the nicotinic acetylcholine receptor.
The image was created by loading the crystal structure of the receptor and
water density on a 3D grid into VMD 1.8.3 and rendered with Raster3D 2.7c.
|
|
|
|
VMD provides the ability to render molecular scenes using
external programs such as ray tracers and commercial
animation packages. This feature can be used to attain
higher image quality than is normally possible using the
built-in OpenGL rendering features in VMD.
The Tachyon parallel ray tracer is distributed with VMD.
|
|
|
|
VMD can display solvent-exluded molecular surfaces through the use
of the program
MSMS.
MSMS provides VMD with fast surface calculation and display, and VMD
uses high-performance display algorithms to allow large or complex
surfaces to be drawn at interactive rates.
|
|
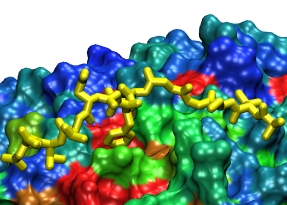
|
|
VMD can now make movies easier than ever before, with the use of a
movie plugin
that takes care of the entire movie making process.
The vmdmovie plugin generates one of several built-in movie types, according
to user selectable options. Once preferences and selections are made, the
movie generator takes control of VMD and takes care of the entire
process, from the generation of individual movie frames using on-screen
snapshots or ray tracers, image format conversion staging of the image
data for compression, invocation of movie compressor programs, and
final disk space cleanup and temporary file deletion. This makes the
whole process of making movies much simpler for inexperienced users.
|
|
|
|
VMD can load and display the results of ab initio simulations done
with packages such as CPMD, GAMESS, Gaussian, ESPRESSO/PWScf, and others.
An excellent tutorial on using VMD with these packages
is available here.
|
|
|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow huge simulation
trajectories to be loaded into physical memory and accomodate large
volumetric datasets. This 400,000 atom virus structure is just a simple
example of what can be done with VMD on an appropriately equipped
graphics workstation.
|
|
|
|
VMD is frequently used to make figures and illustrations that grace
the cover pages of textbooks and journals. A sampling of some of the
VMD cover page images produced by local researchers and collaborators can be
viewed here.
|
|

|
|
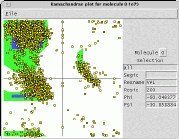
VMD includes a
Ramachandran plot plugin
which plots
Phi and Psi angles for a selected molecule. The angle plot
automatically updates when simulation trajectory frames
are advanced by hand, or when animated. Each plotted angle
is individually selectable, and reports its segment ID,
residue name, residue ID, Phi angle, and Psi angle, when selected.
Ramachandran plots can be saved to a postscript file for inclusion
into publications.
|
|
|
|
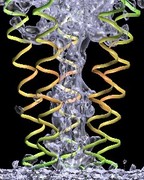
VMD can be used with 3-D printers to create solid models of molecular
structures such as the LH-II
heterodimer unit shown at right. The most recent version of VMD supports
the newest color-capable 3-D printers, and can generate STL or VRML files
suitable for 3-D printing. Two identical LH-II heterodimer units were
printed in 6.4 hours on a Z-Corp Z400
3-D printer at an approximate cost of $1 to $2 per cubic inch.
Each of the two solid models consumed 12.4 cubic inches of the ZP102 powder.
|
|

|
|
VMD provides user-editable "materials" which can be applied to
molecular geometry. These material properties control the
details of how VMD shades the molecular geometry, and how
transparent or opaque the displayed molecular geometry is.
With this feature, one can easily create nice looking transparent
surfaces which allow inner structural details to be seen within a
large molecular structure. The material controls can be particularly
helpful when rendering molecular scenes using external ray tracers,
each of which typically differ slightly.
|
|
|
|
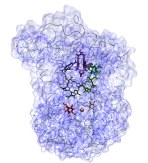
VMD provides a
sequence plugin
which can be used to view a structure's
sequence, secondary structure code, and B value in a scrolling window.
The sequence viewer can zoom in on a region of interest, and selections
can be made on the sequence, which are then highlighted on the structure
in VMD's OpenGL graphics window.
|
|
|
|
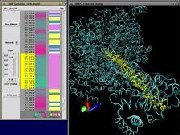
VMD takes advantage of inexpensive game technologies, graphics boards and
stereo glasses to provide capabilities for PC users which were once
only available in $40,000 workstations. For only a few hundred dollars,
it is now possible to equip most desktop PC's with stereoscopic display
capabilities, hardware accelerated 3-D rendering, and six-degree-of-freedom
motion control capability.
|
|

|
|
VMD supports the use of separate displays for its graphical user interface
forms and menus, and its 3-D graphics display window.
This allows VMD to be used easily in a
theatre or auditorium environment
where one display
channel may be projected on a large screen for classes or presentations,
and other screens are only seen by the presenter or helpers. This feature
lets the presenter show just the VMD graphics window on the projection
system, without distracting the audience with other user interfaces,
menus, etc.
|
|

|
|
BioCoRE enhances VMD by making it very
easy to share molecular views with collaborators. Once you have a
desired view in VMD, the view can be saved back to BioCoRE by selecting
an option in a special BioCoRE window. Collaborators can then use
BioCoRE to load the same view in their own copy of VMD.
|
|
|
|
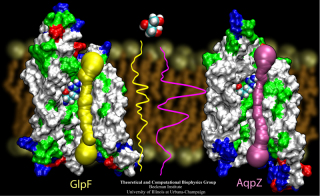
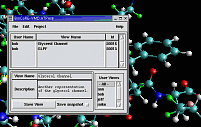
VMD includes built-in support for
IMD (Interactive Molecular Dynamics)
simulations using NAMD, or Protomol.
IMD allows a researcher to interact with molecular structures within
a running simulation, applying forces on groups of atoms in the simulation.
The user experiences force feedback when resistance is encountered, through
the use of 3-D motion control devices with haptic feedback.
IMD techniques are currently being applied to studies of the
Glycerol channel.
|
|
|
|
VMD includes built-in support for high-end quad-buffered
stereoscopic rendering which can be used in projection theatres
as well as desktop graphics workstations. Sterescopic display aids
significantly in the perception of three dimensional structures and
has been a key feature in VMD since its first release. VMD also
provides support for inexpensive game-oriented stereo glasses, and even
anaglyphic (Red/Blue) stereo.
|
|

|
|
VMD can be used with tiled displays and cluster-based rendering
systems for relatively low-cost, high resolution display of
molecular geometry. This feature is particularly attractive for
institutions with tiled display systems used for auditoriums, or
other large-format presentation rooms. The image at the right
shows a six projector tiled display at
NCSA, running VMD.
|
|

|
|
VMD supports the use of six-degree-of-freedom motion control devices
such as the Spaceball and Magellan. These input devices provide an
easy-to-use interface for performing complex 3-D manipulation, and
leave your other hand free to use the mouse and keyboard at the same
time. These devices can also be used to manipulate 3-D pointers
in the scene for meetings and presentations.
|
|

|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow large-size and
long-timescale simulation trajectories to be loaded into physical memory
and accommodate large volumetric datasets.
The 64 million atom HIV capsid simulation
described in the May 30, 2013 issue of Nature is a first-class
example of what can be done with VMD on an appropriately equipped
graphics workstation. The HIV-1 model was prepared for simulation
using the structure building tools and scripting features of VMD were
later used for trajectory analysis. The all-atom structure of the
HIV-1 capsid shown on the Nature cover was rendered within VMD
using the internal
Tachyon
ray tracing engine, and was then composited with an artistic
representation of the viral envelope and the Nature cover text.
|
|
|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow large-size and
long-timescale simulation trajectories to be loaded into physical memory
and accommodate large volumetric datasets.
The 64 million atom HIV capsid simulation
described in the May 30, 2013 issue of Nature is a first-class
example of what can be done with VMD on an appropriately equipped
graphics workstation. The HIV-1 model was prepared for simulation
using the structure building tools and scripting features of VMD were
later used for trajectory analysis. The all-atom structure of the
HIV-1 capsid shown on the Nature cover was rendered within VMD
using the internal
Tachyon
ray tracing engine, and was then composited with an artistic
representation of the viral envelope and the Nature cover text.
|
|
|
|
VMD is capable of working with very large structures up to the limits
of available memory. The 64-bit versions of VMD allow large-size and
long-timescale simulation trajectories to be loaded into physical memory
and accommodate large volumetric datasets.
The 64 million atom HIV capsid simulation
described in the May 30, 2013 issue of Nature is a first-class
example of what can be done with VMD on an appropriately equipped
graphics workstation. The HIV-1 model was prepared for simulation
using the structure building tools and scripting features of VMD were
later used for trajectory analysis. The all-atom structure of the
HIV-1 capsid shown on the Nature cover was rendered within VMD
using the internal
Tachyon
ray tracing engine, and was then composited with an artistic
representation of the viral envelope and the Nature cover text.
|
|
|
|
Human immunodeficiency virus type 1 (HIV-1) is the major cause of
AIDS, for which treatments need to be developed continuously as the
virus becomes quickly resistant to new drugs. When the virus infects
a human cell it releases into the cell its capsid, a closed, stable
container protecting the viral genetic material. However, interaction
with the cell triggers at some point an instability of the capsid,
leading to a well timed release of the genetic material that merges
then with the cell's genes and begins to control the cell. The dual
role of the capsid, to be functionally both stable and unstable, makes
it in principle an ideal target for antiviral drugs and, in fact,
treatments of other viral infections successfully target the
respective capsids. The size of the HIV-1 capsid (about 1,300
proteins), and its irregular shape had prevented so far the resolution
of a full capsid atomic-level structure. However, in a tour de force
effort, groups of experimental and computational scientists have now
resolved the capsid's chemical structure (deposited to the protein
data bank under the accession codes 3J3Q
and 3J3Y).
As reported
recently (see also journal cover), the
researchers combined NMR structure analysis, electron microscopy and
data-guided molecular dynamics
simulations utilizing VMD to prepare
and analyze simulations performed using NAMD on one of the most powerful computers
worldwide, Blue
Waters, to obtain and characterize the HIV-1 capsid. The
discovery can guide now the design of novel drugs for enhanced
antiviral therapy.
More information is available on our virus website, in video, and in a press release.
|
|
See also movie: 15.5MB
made with VMD
|
|
Human immunodeficiency virus type 1 (HIV-1) is the major cause of
AIDS, for which treatments need to be developed continuously as the
virus becomes quickly resistant to new drugs. When the virus infects
a human cell it releases into the cell its capsid, a closed, stable
container protecting the viral genetic material. However, interaction
with the cell triggers at some point an instability of the capsid,
leading to a well timed release of the genetic material that merges
then with the cell's genes and begins to control the cell. The dual
role of the capsid, to be functionally both stable and unstable, makes
it in principle an ideal target for antiviral drugs and, in fact,
treatments of other viral infections successfully target the
respective capsids. The size of the HIV-1 capsid (about 1,300
proteins), and its irregular shape had prevented so far the resolution
of a full capsid atomic-level structure. However, in a tour de force
effort, groups of experimental and computational scientists have now
resolved the capsid's chemical structure (deposited to the protein
data bank under the accession codes 3J3Q
and 3J3Y).
As reported
recently (see also journal cover), the
researchers combined NMR structure analysis, electron microscopy and
data-guided molecular dynamics
simulations utilizing VMD to prepare
and analyze simulations performed using NAMD on one of the most powerful computers
worldwide, Blue
Waters, to obtain and characterize the HIV-1 capsid. The
discovery can guide now the design of novel drugs for enhanced
antiviral therapy.
More information is available on our virus website, in video, and in a press release.
|
|
See also movie: 15.5MB
made with VMD
|
|
Human immunodeficiency virus type 1 (HIV-1) is the major cause of
AIDS, for which treatments need to be developed continuously as the
virus becomes quickly resistant to new drugs. When the virus infects
a human cell it releases into the cell its capsid, a closed, stable
container protecting the viral genetic material. However, interaction
with the cell triggers at some point an instability of the capsid,
leading to a well timed release of the genetic material that merges
then with the cell's genes and begins to control the cell. The dual
role of the capsid, to be functionally both stable and unstable, makes
it in principle an ideal target for antiviral drugs and, in fact,
treatments of other viral infections successfully target the
respective capsids. The size of the HIV-1 capsid (about 1,300
proteins), and its irregular shape had prevented so far the resolution
of a full capsid atomic-level structure. However, in a tour de force
effort, groups of experimental and computational scientists have now
resolved the capsid's chemical structure (deposited to the protein
data bank under the accession codes 3J3Q
and 3J3Y).
As reported
recently (see also journal cover), the
researchers combined NMR structure analysis, electron microscopy and
data-guided molecular dynamics
simulations utilizing VMD to prepare
and analyze simulations performed using NAMD on one of the most powerful computers
worldwide, Blue
Waters, to obtain and characterize the HIV-1 capsid. The
discovery can guide now the design of novel drugs for enhanced
antiviral therapy.
More information is available on our virus website, in video, and in a press release.
|
|
See also movie: 15.5MB
made with VMD
|
|
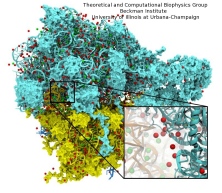
Members of the Theoretical and Computational Biophysics Group
were part of a talented multi-institutional interdisciplinary team awarded
at Supercomputing 2020 with the internationally recognized
ACM Gordon Bell Special Prize for COVID-19 Research,
described as the "Nobel Prize for high performance computing."
The winning team developed a new AI-driven simulation workflow to
drive 8.5M-atom simulations of the SARS CoV-2 spike protein,
its interaction with the human receptor ACE2,
and 305M-atom simulations of the full SARS CoV-2 virion, using
NAMD on
Summit, the most powerful supercomputer in the United States,
operated by the Oak Ridge National Laboratory.
The NAMD simulations collectively performed over one ZettaFLOP
of calculation.
The AI-driven workflow and weighted ensemble methods used by the
team increased effective performance by orders of magnitude.
VMD was used to
build complete structures, prepare the simulations,
analyze the results, and create all of the team's visualizations
due to its unique capacity for working with
extremely large biomolecular systems and diverse structure data.
Due to decades of prior investment in the development of parallel
molecular dynamics, ongoing technology collaborations with
hardware vendors and national laboratories deploying top tier systems,
and our lab's prior work to enable large all-atom simulations of
viruses and other large systems of up to one-billion atoms, NAMD and VMD
were well-prepared for the challenges posed by the project
and its highly compressed research timeline.
The team set several new high-water marks for the full virion simulation,
molecular dynamics strong scaling performance, the
scale of weighed ensemble accelerated sampling, and the
use of AI-driven simulation for multiscale modeling.
This marks the second Gordon Bell Prize for NAMD, which was
also a winner in 2002.
Read more about the Gordon Bell Special Prize for COVID-19 Research here.
|
|
Image courtesy L. Casalino.
Made with VMD
|
|
Members of the Theoretical and Computational Biophysics Group
were part of a talented multi-institutional interdisciplinary team awarded
at Supercomputing 2020 with the internationally recognized
ACM Gordon Bell Special Prize for COVID-19 Research,
described as the "Nobel Prize for high performance computing."
The winning team developed a new AI-driven simulation workflow to
drive 8.5M-atom simulations of the SARS CoV-2 spike protein,
its interaction with the human receptor ACE2,
and 305M-atom simulations of the full SARS CoV-2 virion, using
NAMD on
Summit, the most powerful supercomputer in the United States,
operated by the Oak Ridge National Laboratory.
The NAMD simulations collectively performed over one ZettaFLOP
of calculation.
The AI-driven workflow and weighted ensemble methods used by the
team increased effective performance by orders of magnitude.
VMD was used to
build complete structures, prepare the simulations,
analyze the results, and create all of the team's visualizations
due to its unique capacity for working with
extremely large biomolecular systems and diverse structure data.
Due to decades of prior investment in the development of parallel
molecular dynamics, ongoing technology collaborations with
hardware vendors and national laboratories deploying top tier systems,
and our lab's prior work to enable large all-atom simulations of
viruses and other large systems of up to one-billion atoms, NAMD and VMD
were well-prepared for the challenges posed by the project
and its highly compressed research timeline.
The team set several new high-water marks for the full virion simulation,
molecular dynamics strong scaling performance, the
scale of weighed ensemble accelerated sampling, and the
use of AI-driven simulation for multiscale modeling.
This marks the second Gordon Bell Prize for NAMD, which was
also a winner in 2002.
Read more about the Gordon Bell Special Prize for COVID-19 Research here.
|
|
Image courtesy L. Casalino.
Made with VMD
|
|
Members of the Theoretical and Computational Biophysics Group
were part of a talented multi-institutional interdisciplinary team awarded
at Supercomputing 2020 with the internationally recognized
ACM Gordon Bell Special Prize for COVID-19 Research,
described as the "Nobel Prize for high performance computing."
The winning team developed a new AI-driven simulation workflow to
drive 8.5M-atom simulations of the SARS CoV-2 spike protein,
its interaction with the human receptor ACE2,
and 305M-atom simulations of the full SARS CoV-2 virion, using
NAMD on
Summit, the most powerful supercomputer in the United States,
operated by the Oak Ridge National Laboratory.
The NAMD simulations collectively performed over one ZettaFLOP
of calculation.
The AI-driven workflow and weighted ensemble methods used by the
team increased effective performance by orders of magnitude.
VMD was used to
build complete structures, prepare the simulations,
analyze the results, and create all of the team's visualizations
due to its unique capacity for working with
extremely large biomolecular systems and diverse structure data.
Due to decades of prior investment in the development of parallel
molecular dynamics, ongoing technology collaborations with
hardware vendors and national laboratories deploying top tier systems,
and our lab's prior work to enable large all-atom simulations of
viruses and other large systems of up to one-billion atoms, NAMD and VMD
were well-prepared for the challenges posed by the project
and its highly compressed research timeline.
The team set several new high-water marks for the full virion simulation,
molecular dynamics strong scaling performance, the
scale of weighed ensemble accelerated sampling, and the
use of AI-driven simulation for multiscale modeling.
This marks the second Gordon Bell Prize for NAMD, which was
also a winner in 2002.
Read more about the Gordon Bell Special Prize for COVID-19 Research here.
|
|
Image courtesy L. Casalino.
Made with VMD
|
|
Sunlight powers life on Earth. This basic fact has been known since ancient times and retold many times in many cultures. Today, scientists understand the means through which light powers life at atomistic detail, as a chain of processes climbing scales from electronic interactions to cooperation between proteins to cell-scale integration of energy conversion. These processes have been illustrated in a recent video
that was awarded the
BEST SCIENTIFIC VISUALIZATION OF 2019
at the SC19 conference, Scientific Visualization & Data Analytics Showcase.
The video is a joint production with the team of
Donna Cox
at
NCSA
and is based on a decade long collaborative effort with the experimental group of
Neil Hunter
and a lifetime research interest for
Klaus Schulten.
The underlying scientific investigation illustrated in the video
was presented in
18 manuscripts over the past decade,
from
atomic scale structural modeling
to
organelle-scale
and
cell-scale
integration of function.
These modeling efforts also led to
a molecular dynamics simulation of the chromatophore
using NAMD,
recently published in
Cell.
The video segment,
produced with VMD,
that won as the Best Scientific Visualization is an excerpt from
the fulldome movie
'Birth of Planet Earth' released to planetariums worldwide by Spitz Creative Media.
The oldest story of humanity — light powering life — coming soon to a theater near you.
|
|
image size: 621K
made with VMD
|
|
Sunlight powers life on Earth. This basic fact has been known since ancient times and retold many times in many cultures. Today, scientists understand the means through which light powers life at atomistic detail, as a chain of processes climbing scales from electronic interactions to cooperation between proteins to cell-scale integration of energy conversion. These processes have been illustrated in a recent video
that was awarded the
BEST SCIENTIFIC VISUALIZATION OF 2019
at the SC19 conference, Scientific Visualization & Data Analytics Showcase.
The video is a joint production with the team of
Donna Cox
at
NCSA
and is based on a decade long collaborative effort with the experimental group of
Neil Hunter
and a lifetime research interest for
Klaus Schulten.
The underlying scientific investigation illustrated in the video
was presented in
18 manuscripts over the past decade,
from
atomic scale structural modeling
to
organelle-scale
and
cell-scale
integration of function.
These modeling efforts also led to
a molecular dynamics simulation of the chromatophore
using NAMD,
recently published in
Cell.
The video segment,
produced with VMD,
that won as the Best Scientific Visualization is an excerpt from
the fulldome movie
'Birth of Planet Earth' released to planetariums worldwide by Spitz Creative Media.
The oldest story of humanity — light powering life — coming soon to a theater near you.
|
|
image size: 621K
made with VMD
|
|
Modern molecular simulation, visualization, and modeling techniques often require high-performance computing hardware
to obtain the best efficiency. However, access to such hardware can be a barrier for some researchers.
As an alternative, cloud computing provides a cost-effective and practical solution for many molecular modeling tasks and for small and moderate size molecular dynamics simulations. The cloud computing model
provides researchers with access to powerful computational equipment
that would otherwise be too costly to procure, maintain,
and administer on their own. Additionally, cloud platforms can easily bundle different software packages
used in a modeling workflow to guaruntee their availablity and
interoperability on a standardized system.
We have adapted our molecular modeling applications NAMD, VMD, and associated tools to
operate within the Amazon Web Services (AWS) Elastic Compute Cloud (EC2) platform, enabling popular
research workflows to be run remotely by scientists all over the globe,
with no need for investment in local computing resources and
a reduced requirement for expertise in high performance computing technologies.
Recent advancements in GPU virtualization technology now make it possible to use cloud computing for
large-scale scientific computing, data analysis, and visualization tasks. The latest release of our
cloud software can be run on EC2's latest graphics hardware,
backed by DCV, to provide a smooth and seamless
graphical working environment. Instance types specially designed for parallel CPU or GPU workloads
provide users with access to the hardware they need to run even the most demanding features of NAMD and VMD,
all from their much less powerful personal computers and even laptops. These instances can be paid for on an on-demand, as-needed basis, and some
workflows, such as Molecular Dynamics Flexible Fitting, can be run for less than the cost of a cup of coffee.
Our cloud virtual machine image is availble in the Amazon Marketplace,
which provides users with
a very simple one-click launch using pre-configured choices
of instance types that have been tested with our software.
To learn more,
see our
cloud research page.
|
|
|
|
Modern molecular simulation, visualization, and modeling techniques often require high-performance computing hardware
to obtain the best efficiency. However, access to such hardware can be a barrier for some researchers.
As an alternative, cloud computing provides a cost-effective and practical solution for many molecular modeling tasks and for small and moderate size molecular dynamics simulations. The cloud computing model
provides researchers with access to powerful computational equipment
that would otherwise be too costly to procure, maintain,
and administer on their own. Additionally, cloud platforms can easily bundle different software packages
used in a modeling workflow to guaruntee their availablity and
interoperability on a standardized system.
We have adapted our molecular modeling applications NAMD, VMD, and associated tools to
operate within the Amazon Web Services (AWS) Elastic Compute Cloud (EC2) platform, enabling popular
research workflows to be run remotely by scientists all over the globe,
with no need for investment in local computing resources and
a reduced requirement for expertise in high performance computing technologies.
Recent advancements in GPU virtualization technology now make it possible to use cloud computing for
large-scale scientific computing, data analysis, and visualization tasks. The latest release of our
cloud software can be run on EC2's latest graphics hardware,
backed by DCV, to provide a smooth and seamless
graphical working environment. Instance types specially designed for parallel CPU or GPU workloads
provide users with access to the hardware they need to run even the most demanding features of NAMD and VMD,
all from their much less powerful personal computers and even laptops. These instances can be paid for on an on-demand, as-needed basis, and some
workflows, such as Molecular Dynamics Flexible Fitting, can be run for less than the cost of a cup of coffee.
Our cloud virtual machine image is availble in the Amazon Marketplace,
which provides users with
a very simple one-click launch using pre-configured choices
of instance types that have been tested with our software.
To learn more,
see our
cloud research page.
|
|
|
|
On April 20th, 2017, the Journal of Physical Chemistry published a Memorial Issue in honor of Klaus Schulten gathering more than 60 research articles. Klaus was an undisputed leader in theoretical and computational biophysics, recognized by his peers for his immense contribution to the field, and having devoted his entire career to establish the mechanisms that underlie cellular processes using the laws of physics. Originally planned to be a Festschrift to celebrate Klaus's achievements on his 70th birthday, the Memorial Issue initiative immediately triggered a unanimous positive response from friends, academic colleagues and longtime collaborators across the world. The many contributions assembled in the Memorial Issue of the Journal of Physical Chemistry lie at the confluence of theory and experiment, and cover a broad gamut of topics that were dear to Klaus, ranging from photosynthesis to molecular machines and membrane proteins. We gratefully acknowledge the many authors of the Memorial Issue, who enthusiastically accepted to pay one last homage to Klaus through contributions of very high scientific quality.
|
|
image size:
213K
|
|
On April 20th, 2017, the Journal of Physical Chemistry published a Memorial Issue in honor of Klaus Schulten gathering more than 60 research articles. Klaus was an undisputed leader in theoretical and computational biophysics, recognized by his peers for his immense contribution to the field, and having devoted his entire career to establish the mechanisms that underlie cellular processes using the laws of physics. Originally planned to be a Festschrift to celebrate Klaus's achievements on his 70th birthday, the Memorial Issue initiative immediately triggered a unanimous positive response from friends, academic colleagues and longtime collaborators across the world. The many contributions assembled in the Memorial Issue of the Journal of Physical Chemistry lie at the confluence of theory and experiment, and cover a broad gamut of topics that were dear to Klaus, ranging from photosynthesis to molecular machines and membrane proteins. We gratefully acknowledge the many authors of the Memorial Issue, who enthusiastically accepted to pay one last homage to Klaus through contributions of very high scientific quality.
|
|
image size:
213K
|
|
On April 20th, 2017, the Journal of Physical Chemistry published a Memorial Issue in honor of Klaus Schulten gathering more than 60 research articles. Klaus was an undisputed leader in theoretical and computational biophysics, recognized by his peers for his immense contribution to the field, and having devoted his entire career to establish the mechanisms that underlie cellular processes using the laws of physics. Originally planned to be a Festschrift to celebrate Klaus's achievements on his 70th birthday, the Memorial Issue initiative immediately triggered a unanimous positive response from friends, academic colleagues and longtime collaborators across the world. The many contributions assembled in the Memorial Issue of the Journal of Physical Chemistry lie at the confluence of theory and experiment, and cover a broad gamut of topics that were dear to Klaus, ranging from photosynthesis to molecular machines and membrane proteins. We gratefully acknowledge the many authors of the Memorial Issue, who enthusiastically accepted to pay one last homage to Klaus through contributions of very high scientific quality.
|
|
image size:
213K
|
Back to the main page