Virus Spotlights
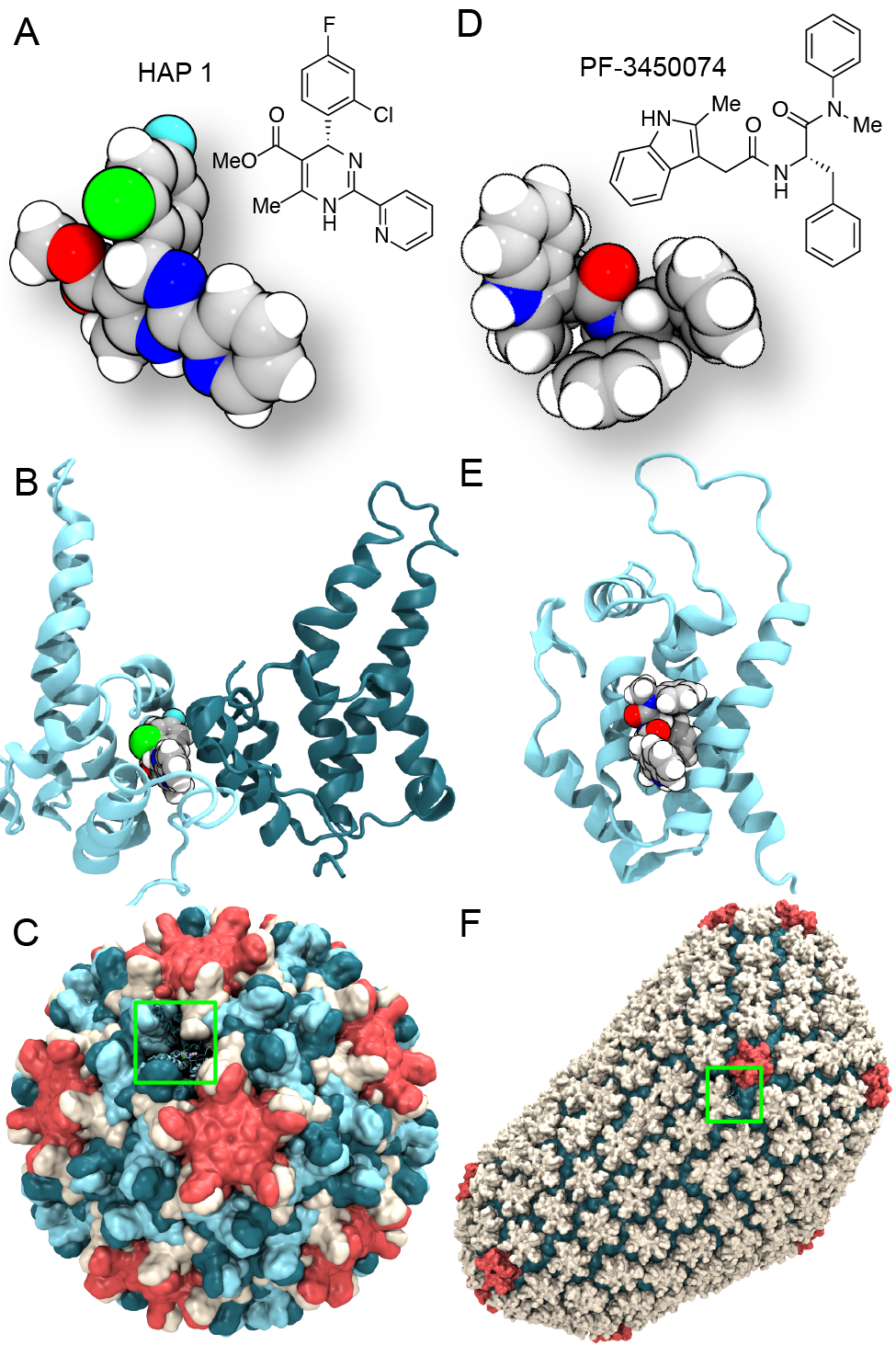
image size:
0 bytes
made with VMD
Virus capsids, specialized protein shells that encase the genome of viral pathogens, play critical roles in regulating viral infection, and are, thus, of great pharmacological interest as drug targets. In particular, small-molecule drugs (typically <900 Da) represent a promising antiviral strategy, and a number of such compounds have been developed to inhibit virus capsids by interfering with their assembly and uncoating processes. Importantly, to explain the mechanisms by which drugs disrupt capsid function, as well as to successfully design novel therapeutics, the interactions of drug molecules with their capsid targets must be studied at full chemical detail. Toward this end, all-atom molecular dynamics simulations are emerging as an essential technique to investigate the effects of small-molecule drugs on capsid structure and dynamics. Research presented in a recent Perspective applying simulations in NAMD to study drug-bound hepatitis B virus (HBV) and human immunodeficiency virus type 1 (HIV-1) capsids suggests the types of valuable chemical and physical information computational approaches can reveal, and underscores the importance of simulating, not isolated capsid proteins, but functional assemblies up to the level of complete capsids. Notably, through analysis with VMD, the study found that binding of the drug HAP1 to the HBV capsid causes global structural changes that subtly alter the overall capsid shape, including a flattening of capsid curvature. Further, the study found that the binding of the drug PF74 to the HIV-1 capsid imposes rigidity and causes shifts in allosteric communication pathways connecting distant regions of the capsid protein. The authors of the Perspective anticipate that many other such exciting discoveries regarding virus capsid function and their use as drug targets lie just ahead on the horizon, and molecular dynamics simulations will drive these discoveries pending a series of notable advancements in computational methodology.

image size:
1.7MB
made with VMD
Our lungs are coated with a layer of protein and lipid mixture called lung surfactant, which prevents the lungs from collapsing and protects us from bacterial and viral infections (see October 2012 and January 2014 highlights). Lung surfactant protein A (SP-A) - the major protein constituent of lung surfactant - plays a dual role. It aggregates DPPC lipid, a major component of lung membrane, into a lattice-like structure that prevents the lungs from collapsing. SP-A is also known to recognize and bind bacterial lipids, namely lipid A, on surfaces of gram-negative bacteria, thereby helping to initiate various clearance mechanisms. However, it was unclear how SP-A exhibits such functional duality with its binding to two different types of lipids. A recent study used molecular dynamics simulations with NAMD to unravel the dual role of SP-A. Combined with crystallographic and mutational analyses, researchers have discovered several critical, non-canonical lipid binding sites that involves cation-π interactions and hydrogen bonds. Simulations have also revealed that SP-A binds stronger to bacterial lipid (lipid A) than to surfactant lipid (DPPC lipid), which suggests SP-A may prioritize its host defense functions by transferring from lung membrane to bacterial surface. These findings in atomistic detail will enable experimentalists to enhance the antimicrobial function of SP-A. More on our lung surfactant protein website.
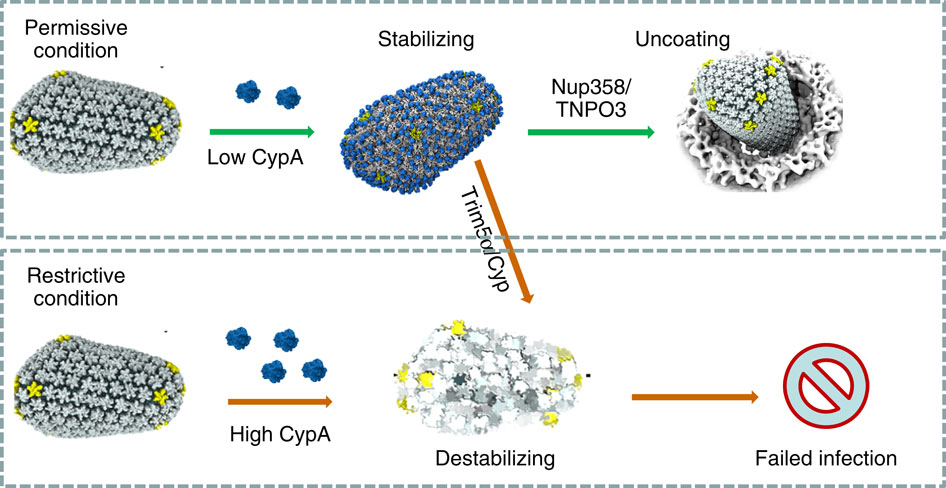
image size:
101.5KB
made with VMD
When experimental-computational biologists embarked on the great challenge of resolving the atomic level structure of the HIV virus capsid that contains the virus' deadly genetic cargo, they were advised by referees not to try as the capsid is too big, too irregular, and nobody would need the highly resolved structural information. Stubbornly, the researchers went ahead anyway and succeeded getting the atomic resolution structure, overcoming size and irregularity challenges (see highlight Elusive HIV-1 Capsid). But the question remained: Is the atomic level structure of the huge HIV capsid made of 1,300 proteins useless? The HIV capsid is a closed container made of protein pentamers and hexamers, with a surface of continuously changing curvature. Two recent experimental-computational studies demonstrate now that the capsid structure is far from useless, in fact, it is a great treasure. The first study was published last year and showed that the human protein, Cyclophilin-A (CypA), involved in several diseases, interacts with the HIV capsid and affects the capsid's dynamic properties (see highlight HIV, Cells and Deception). In a second, recent study, guided by cryo-EM measurements and benefiting from large-scale molecular dynamics simulations with NAMD, researchers could resolve with new accuracy the binding of hundreds of CypA proteins on the capsid's surface. They found that CypA binds along high curvature lines of the capsid, which enhances stiffness and stability of the capsid, even though only about half of the capsid is actually covered by CypA. The limited levels of CypA stabilize and protect the viral capsid as it moves through the infected cell towards the cell's nuclear pore where nuclear proteins additionally bind to the capsid at places not covered by CypA and promote there uncoating and release of the capsid cargo into the nucleus. More information is available on our retrovirus website and in a news release.
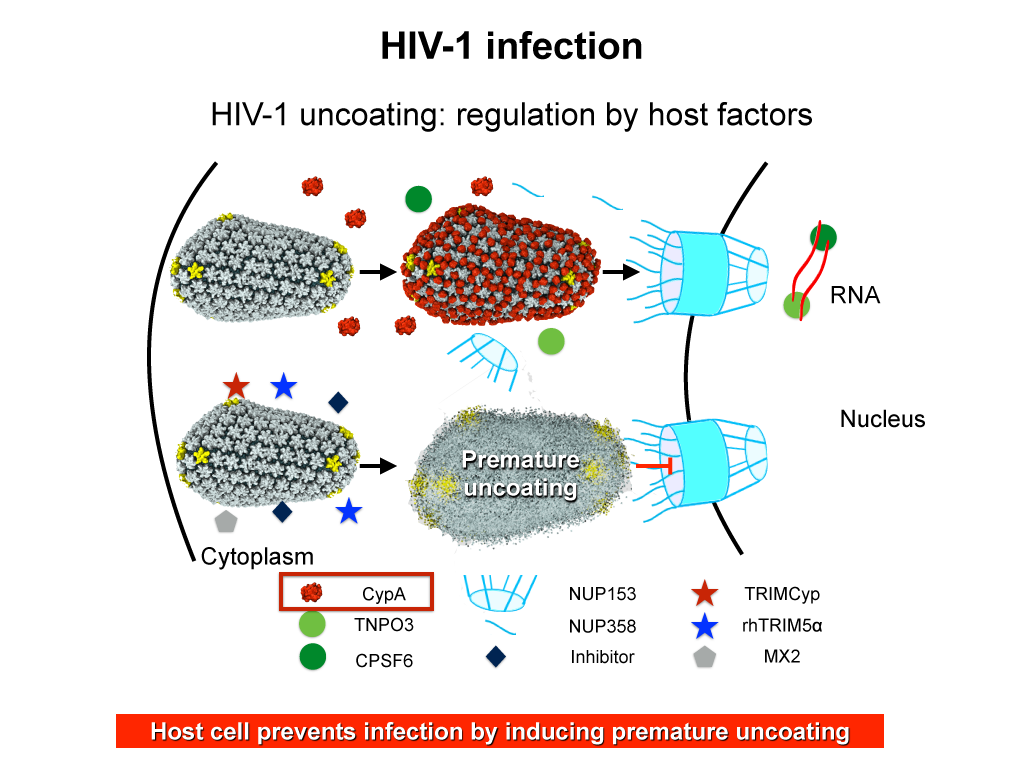
image size:
283.9KB
made with VMD
When human immundeficiency virus (HIV) infects a human cell, it releases into the interior of the cell its capsid (made of about 1,300 identical so-called CA proteins), a closed, stable container that protects the viral genetic material (see also June 2013 highlight Elusive HIV-1 Capsid and August 2015 highlight Anatomy of a Dormant Killer). Once in the cell ― while avoiding detection by cellular proteins ― the capsid deceives the cell and directs the cell machinery to transport it to the nucleus. The human-cell protein Cyclophilin A (CypA) is thereby exploited to act against the cell's well being and to assist the HIV infection by getting the capsid to access the cell nucleus; this results in a delicate choreography accomplished by escaping anti-viral proteins in the cell and deceiving transport proteins at the nucleus, all of which contain a CypA domain that interacts directly with the capsid. Despite the availability of the crystal structure of the complex of CypA and CA proteins determined nearly 20 years ago, the mechanism by which CypA assists the capsid has been unclear due to the lack of information on CypA in complex with not one CA protein, but the entire capsid. In collaboration with experimental groups, computational biologist have shown in a recent report that the effects of CypA on the capsid are not only structural, but also dynamical. Thus, new therapeutic strategies may be envisioned through modulation of the dynamics of the capsid by small-molecule (drug) compounds that inhibit the binding of CypA to the capsid. More information is available on our retrovirus website and in a YouTube video.
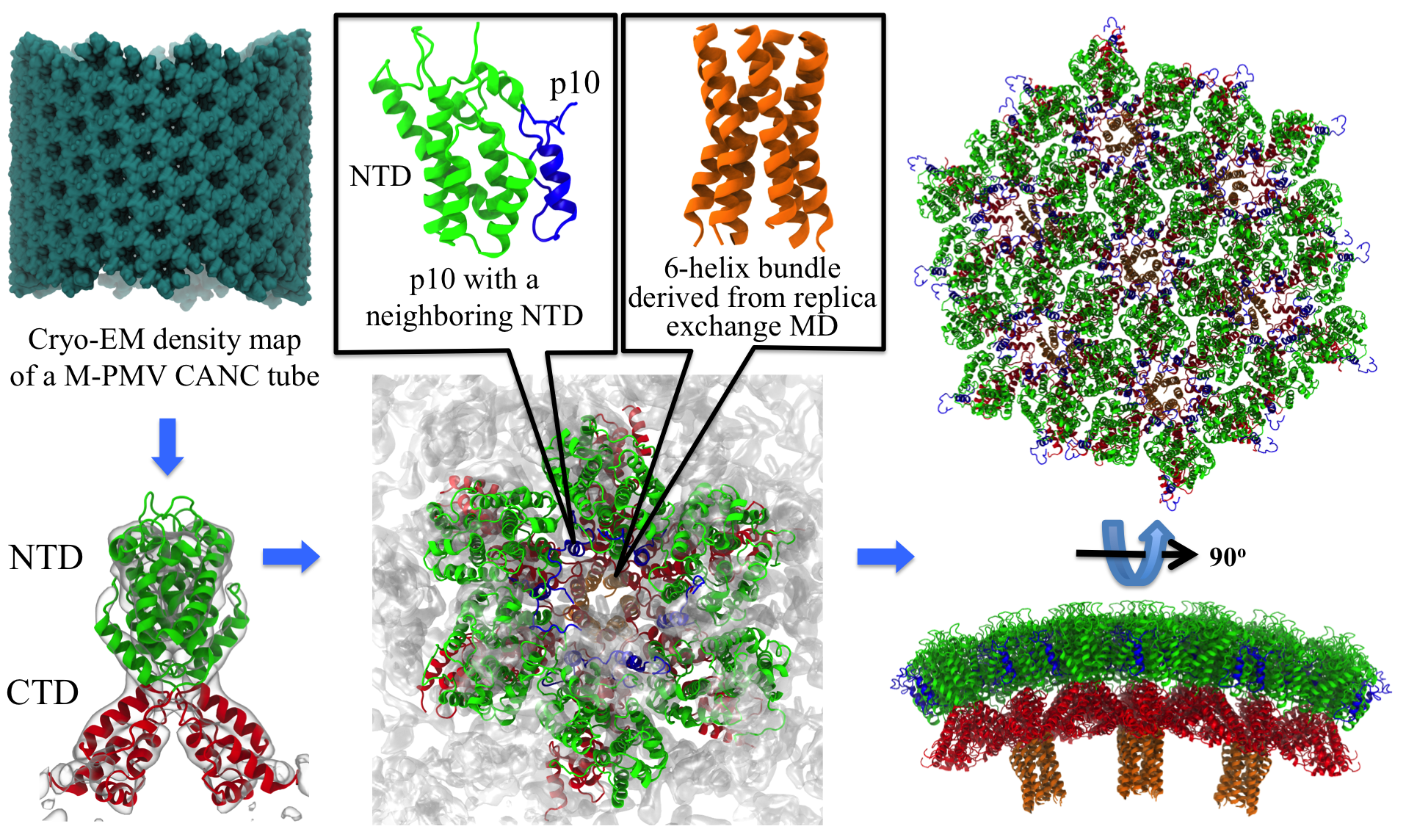
image size:
2.6MB
made with VMD
Retroviruses are parasites that pose a major health threat to humans (for example in case of HIV) and other animals (for example in case of RSV, M-PMV, MLV, and many more viruses). After a retrovirus hijacks a cell, the infected cell produces multiple copies of the virus which are then released into the host's bloodstream. These newly released viruses must mature before they can infect other cells. A strategy for preventing virus spread is therefore, to lock the viral particles in their immature, non-infectious state. However, to render the immature virus an attractive target for structure-based drug development one needs to know its chemical structure. Unfortunately, the complexity and size of the viral particle ― an incomplete hexagonal shell with a size close to 100 nm ― have prevented the experimental determination of the chemical, namely atomic level, structure of the virus. As reported recently, a team of computational and experimental researchers have provided an atomic structure of the immature retroviral lattice for the Rous Sarcoma Virus. The multi-domain RSV model was derived through a combination of state-of-the-art modeling techniques, including, cryo-EM-guided homology modeling, large-scale molecular dynamics simulations using enhanced sampling capabilities available in NAMD, together with experimental measurements such as X-ray crystallography and a wealth of biochemical data. Particularly, the model reveals novel features of the packing and dynamics of the immature capsid protein with implications for the maturation process and confirms the stabilizing roles of the so-called upstream and downstream domains of the immature RSV. More information is available on our retrovirus website, and in a highlight video.
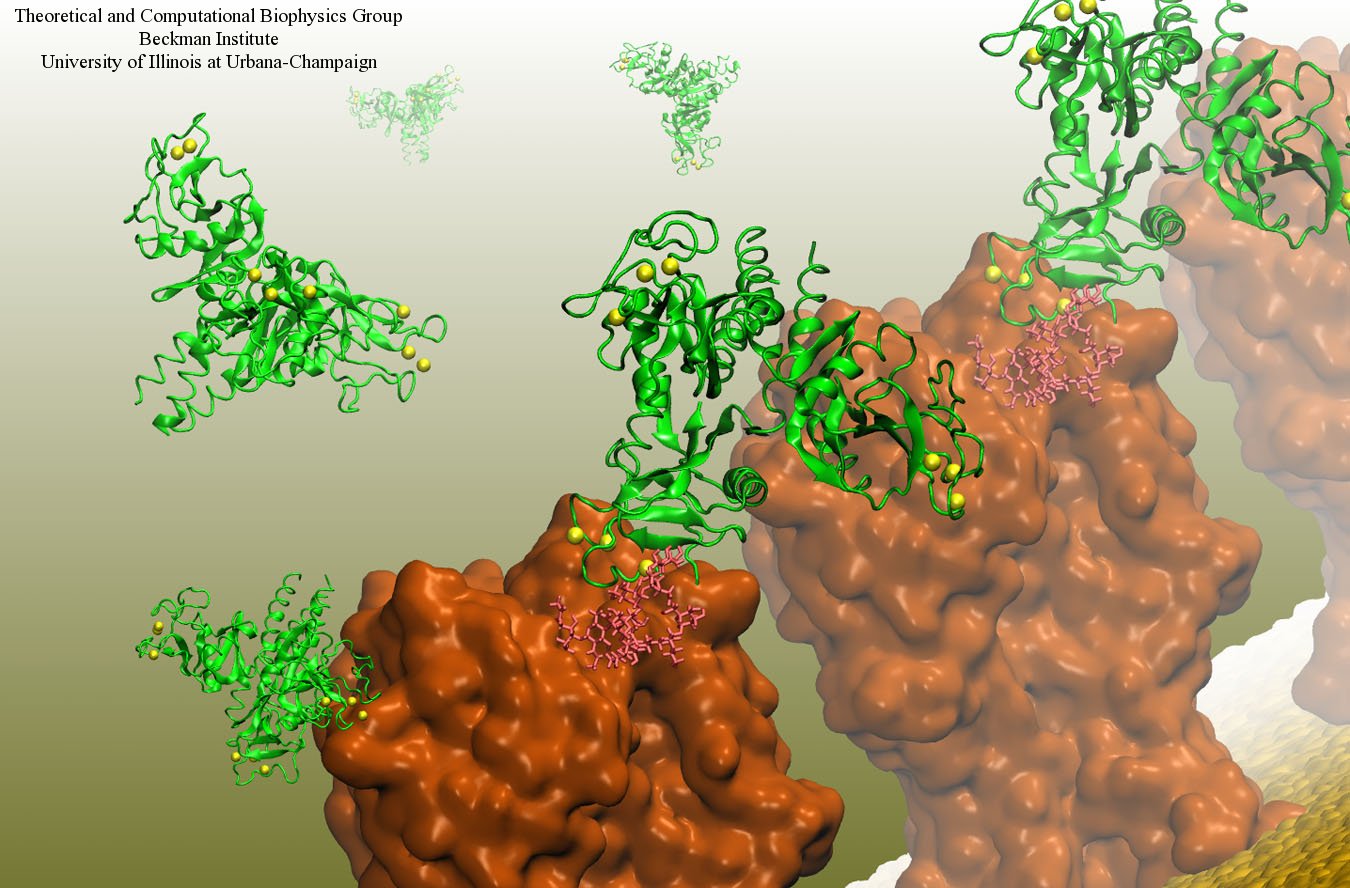
image size:
199.7KB
made with VMD
Our body uses several defense mechanisms against seasonal flu, the common affliction caused by influenza viruses. By taking a yearly flu shot, our body's defense based on antibodies is trained and envoked. A defense system not based on antibodies acts at the very front line of influenza virus attack, namely the lungs. For this protection the body uses so-called lung surfactant proteins that coat the inner lining of the lungs to keep a wet film on the lung surface needed for oxygen-carbon dioxide exchange. The lung surfactant proteins also serve as police against influenza viruses. For this purpose the lung surfactant protein D (SP-D) recognizes a protein component of the virus surface, namely hemagglutinin, and handcuffs the sugar molecules bound to hemagglutinin. A previous experimental-simulation study (see October 2012 highlight) found that SP-D of pigs exhibits a stronger inhibitory activity against influenza A virus in this regard than does human SP-D. In a recent study, researchers have now boosted the protective ability of human SP-D by introducing mutations. Molecular dynamics simulations using NAMD suggest that the mutated human SP-D employs a different and stronger blocking mechanism on the active site of influenza A virus than native SP-D does. Combined with experimental results, the simulations suggest a mechanism through which SP-D acts, namely, by handcuffing viruses together and, thereby, preventing viral entry into cells. The findings from this research might lead to a new protection against seasonal flu, namely a nasal spray containing mutated lung surfactant proteins that strengthen a person's armada of defense proteins on the lung surface. More on our lung surfactant protein website.
Human immunodeficiency virus type 1 (HIV-1) is the major cause of AIDS, for which treatments need to be developed continuously as the virus becomes quickly resistant to new drugs. When the virus infects a human cell it releases into the cell its capsid, a closed, stable container protecting the viral genetic material. However, interaction with the cell triggers at some point an instability of the capsid, leading to a well timed release of the genetic material that merges then with the cell's genes and begins to control the cell. The dual role of the capsid, to be functionally both stable and unstable, makes it in principle an ideal target for antiviral drugs and, in fact, treatments of other viral infections successfully target the respective capsids. The size of the HIV-1 capsid (about 1,300 proteins), and its irregular shape had prevented so far the resolution of a full capsid atomic-level structure. However, in a tour de force effort, groups of experimental and computational scientists have now resolved the capsid's chemical structure (deposited to the protein data bank under the accession codes 3J3Q and 3J3Y). As reported recently (see also journal cover), the researchers combined NMR structure analysis, electron microscopy and data-guided molecular dynamics simulations utilizing VMD to prepare and analyze simulations performed using NAMD on one of the most powerful computers worldwide, Blue Waters, to obtain and characterize the HIV-1 capsid. The discovery can guide now the design of novel drugs for enhanced antiviral therapy. More information is available on our virus website, in video, and in a press release.

Rabbit hemorrhagic disease is extremely contagious and associated with liver necrosis, hemorrhaging, and high mortality in adult rabbits. First described in China in 1984, within a few years, rabbit hemorrhagic disease spread to large parts of the world and today threatens the rabbit industry and related ecology. The disease is caused by a virus, aptly named rabbit hemorrhagic disease virus. As reported recently, a group of experimental and computational researchers combining crystallography, electron microscopy and data-guided molecular dynamics simulations utilizing NAMD determined an atomic model of the capsid, namely the protein shell that surrounds the genetic material of the virus. The capsid simulations involved 10 million atoms and have become feasible only through Blue Waters, a brand new petascale supercomputer. The atomic model, analyzed by means of VMD, recently adapted to studies of very large structures, resolves the structural framework that furnishes both mechanical protection to the viral genes as well as a quick release mechanism after a virus enters a host cell. Researchers can use the detailed knowledge of the capsid structure to develop vaccines against rabbit hemorrhagic disease. More information is available on our virus website and in a news story.

image size:
77.5KB
made with VMD
Did you get your flu shot this year? Influenza is a leading cause of preventable death in the industrialized world, representing hundreds of billions of dollars in healthcare expenditures and loss of economic production. While the yearly influenza vaccination is nearly 90% effective at limiting infections in populations less than 65 years of age, there is insufficient evidence regarding the effectiveness of the flu shot for the elderly population, whose immune systems may not mount an adequate antibody response to vaccination. Beyond vaccination, front-line therapies such as the neuraminidase inhibitors Tamiflu and Relenza have proven to be of limited effectiveness due to the evolution of drug-resistant influenza mutants. Therefore, a need exists for the development of new therapies to circumvent these resistance mechanisms. Computational biologists employing NAMD and VMD used molecular simulations to uncover the key role that water plays in mediating how well antiviral drugs can bind to proteins of the influenza virus. This investigation, reported recently, reveals that amino-acid mutations responsible for drug resistance act by reshaping the local electric field and also by permitting infiltration of water within the otherwise hydrophobic drug binding pocket. These mutations thus induce drug resistance in much the same way as inverting the polarity of a magnet can repel rather than attract. These findings are expected to help guide the design of novel drugs with increased antiviral efficacy. Additional details about this study can be found here.
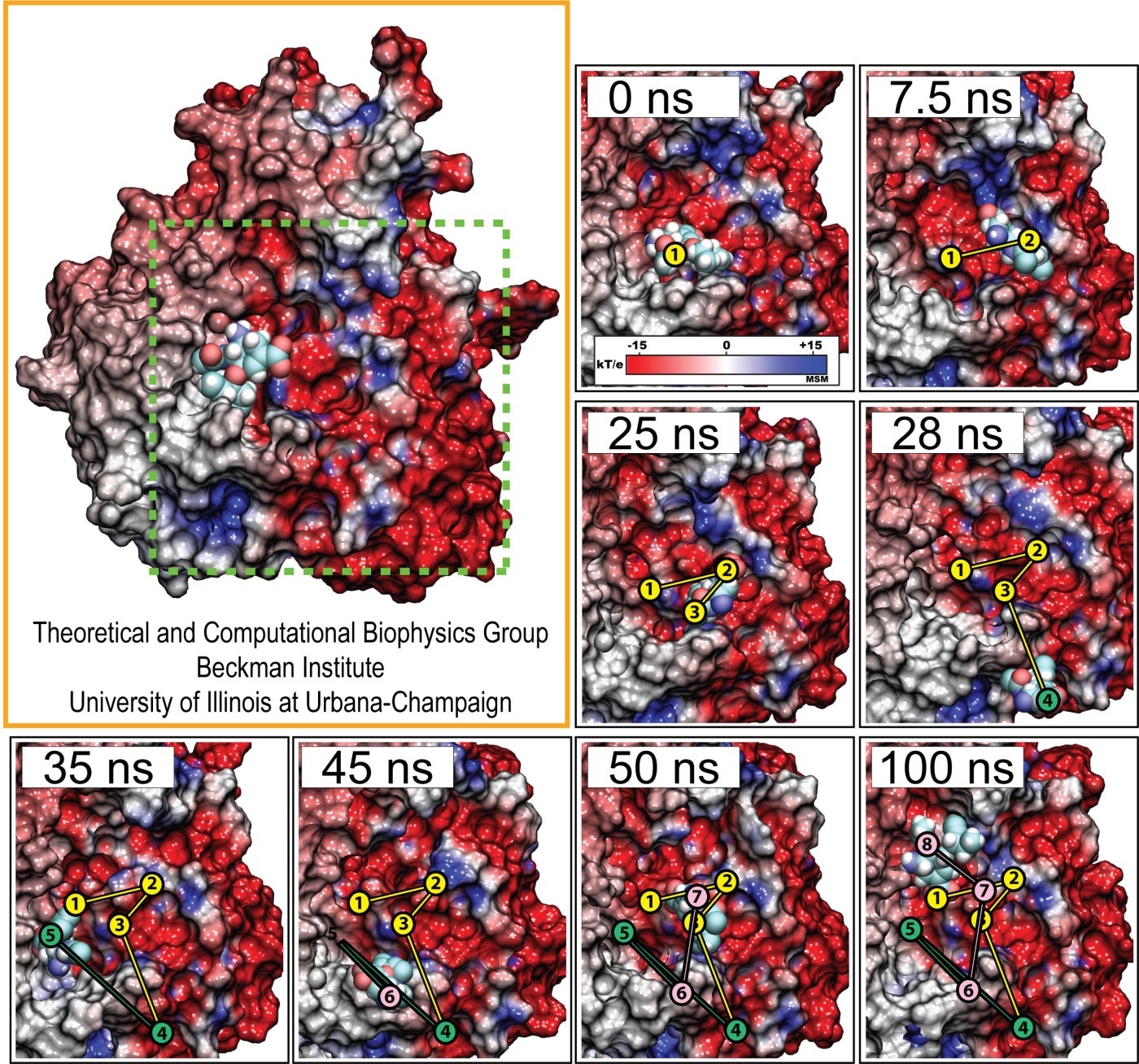
Fever, chills, sore throat, coughing, aches, and pains? Ah ..... you have the flu! As a measure of prevention, vaccines against seasonal influenza are distributed and administered each fall. Last year though, the outbreak of the H1N1pdm "swine" influenza virus, caught health workers by surprise as this virus not only infected individuals during the spring and summer months, but also seemed to be particularly virulent towards otherwise healthy young people. Even more alarming was increasing evidence that H1N1pdm had acquired resistance to the frontline antiflu drug, Tamiflu. In response to this, computational biologists at the University of Illinois and the University of Utah teamed up to uncover the basis for influenza drug resistance through quantum chemistry, and molecular dynamics simulations with NAMD. The results of this study have recently been reported, and uncovered a two stage binding pathway for Tamiflu in H1N1pdm "swine" and H5N1 "avian" flu proteins, as well as a possible mechanism through which genetic mutations can induce drug resistance in one of the stages. Subsequent efforts at drug design against influenza can take advantage of this discovery. This discovery was made possible through use of so-called GPU computing (see Oct 2007 highlight "Graphics Processors Speed Up Simulations"). More information can be found here.
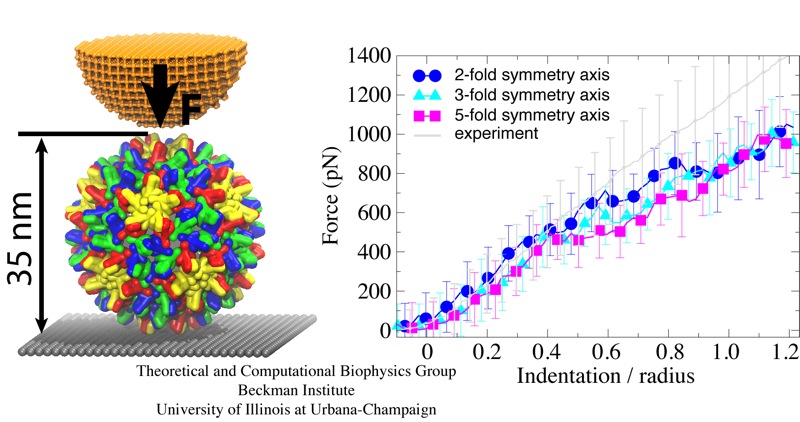
Viruses are the simplest life forms known. In fact, one can question if they are life forms at all, as they cannot exist without infecting a host cell and using its machinery for replication. The virus is indeed just a package material surrounding a genetic message that instructs the host cell to replicate the virus. It looks like a soccer ball, but is a million times smaller (see also the March 2006 and January 2007 highlights). The infection, a well known example being infection of human cells by a flu virus, involves the virus to approach a human cell and dock onto it, become internalized by the cell, bursting then its package, called the capsid, and release the genetic message. The virus capsid needs to be sturdy and impermeable up to the approach to the cell, but then become brittle and porous to release the genetic material. Obviously, the virus capsid must have very distinct mechanical properties to function. To investigate these properties experimental and computational biophysicists teamed up. The experimentalists placed empty capsids of the hepatitis B virus onto a small chip and mechanically squeezed the capsid then with an extremely small tip, measuring how much force is needed to squeeze the spherical capsid down repeatedly. Computational researchers using NAMD repeated the experiment in simulation. As they reported recently, simulation gave the same forces as the experiment, but yielded also a detailed picture of the capsid mechanics. More on our "Molecular dynamics of viruses" web site.
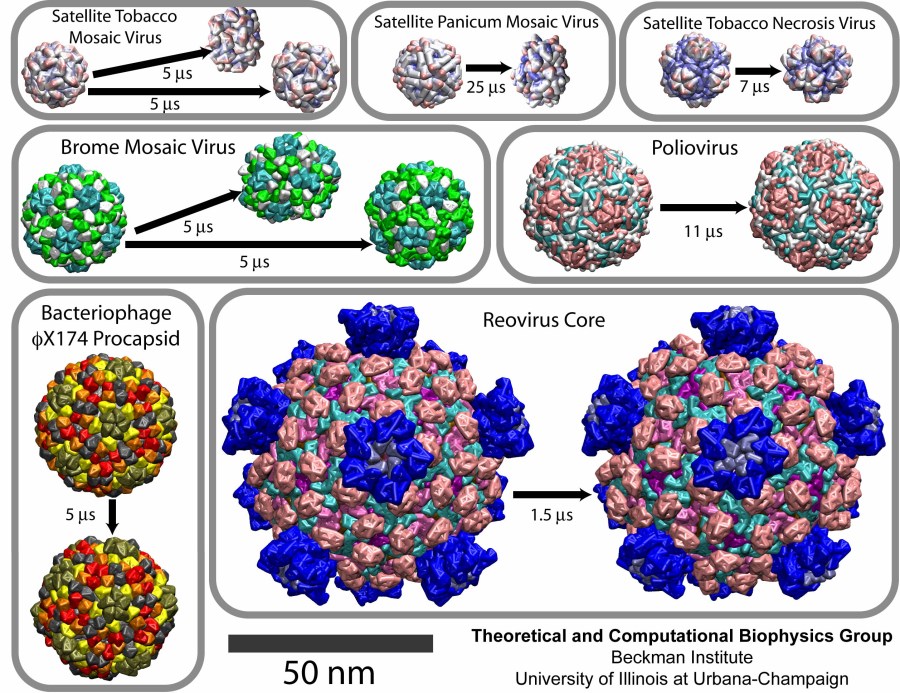
image size:
219.5KB
made with VMD
Viruses are the cause of many human diseases, from the common cold to AIDS, and medicine is continuously searching for better ways to battle viruses through vaccination or medication. Detailed knowledge of the life cycles of viruses should be useful in the treatment of viral diseases. A key focus of investigations is the virus capsid, a protein coat that protects the viral genome, but also triggers release of the genome and other viral factors upon contact with the body's cells. X-ray crystallography has resolved the average structures of many types of virus capsids, providing the basis for detailed investigations, for example by means of molecular dynamics methods, of capsid dynamical properties, e.g., in assembly and disassembly. Unfortunately, due to their large size most virus capsids are beyond the reach of molecular dynamics simulations, with one notable exception (see the March 2006 highlight "Simulating an Entire Life Form"). This earlier simulation allowed researchers to develop and test a method for coarse-grained molecular dynamics simulations that glosses over atomic detail and, thereby, permits microsecond descriptions of entire viral particles. As reported recently (see also journal cover) such simulations, employing the program NAMD, were applied to the empty capsids of several viruses. These simulations revealed a variety of behaviors, from rapid collapse to high stability, depending on the strength of interactions between the proteins from which capsids are built. The new method offers unprecedented views of capsid dynamics that may assist in battling viral diseases. More information on the simulations can be found on our virus web page.
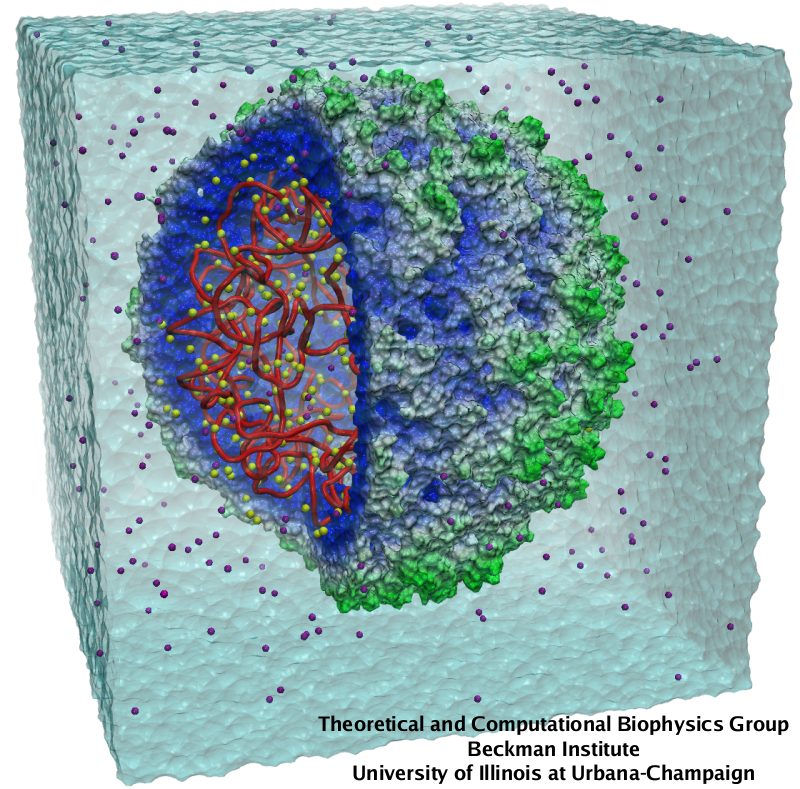
image size:
150.4KB
made with VMD
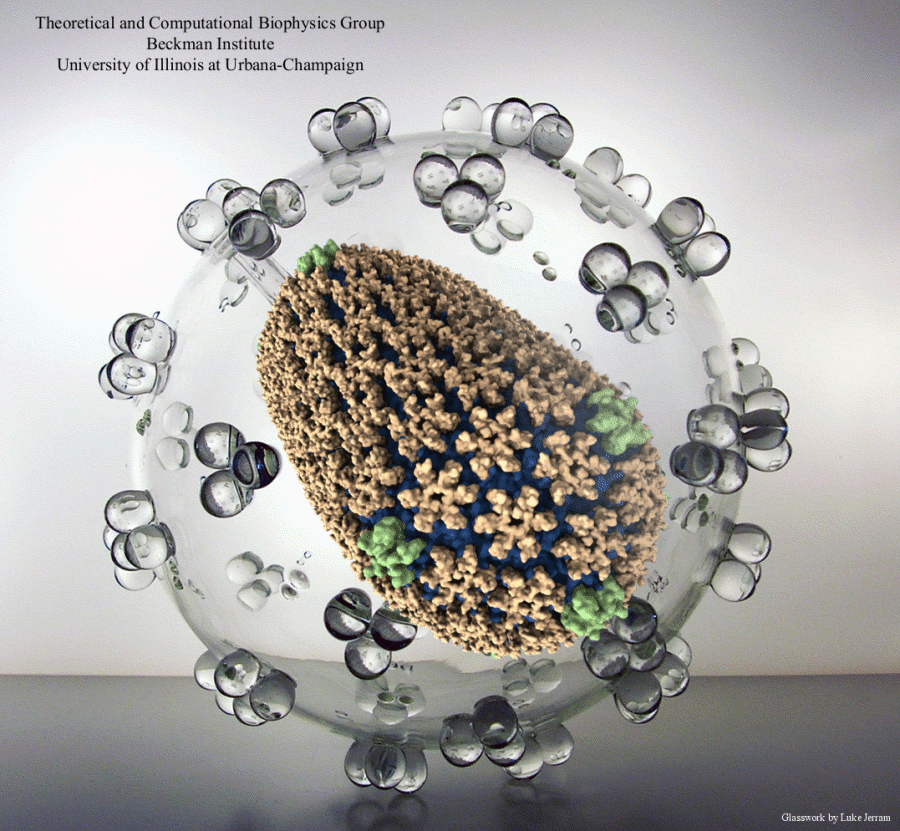
Viruses, the cause of many diseases, are the smallest natural organisms known. They are extremely primitive and parasitic such that biologists refer to them as "particles", rather than organisms. Viruses contain in a protein shell, the capsid, their own building plan, the genome, in the form of DNA or RNA. Viruses hijack a biological cell and make it produce from one virus many new ones. Viruses have evolved elaborate mechanisms to infect host cells, to to produce and assemble their own components, and to leave the host cell when it bursts from viral overcrowding. Because of their simplicity and small size, computational biologists selected a virus for their first attempt to reverse-engineer in a computer program, NAMD, an entire life form, choosing one of the tiniest viruses for this purpose, the satellite tobacco mosaic virus. As described in a recent report, the researchers simulated the virus in a small drop of salt water, altogether involving over a million atoms. This provided an unprecedented view into the dynamics of the virus for a very brief time, revealing nevertheless the key physical properties of the viral particle as well as providing crucial information on its assembly. It may take still a long time to simulate a dog wagging its tail in the computer, but a big first step has been taken to simulate living organisms. Naturally, this step will assist modern medicine (more on our satellite tobacco mosaic virus web page).



{kind=link}