VMD-L Mailing List
From: Jan Saam (saam_at_charite.de)
Date: Wed Jan 31 2007 - 14:30:55 CST
- Next message: ivana adamovic: "Re: About CHARMM electrostaic potential map"
- Previous message: Jan Saam: "Re: Regarding paratool"
- In reply to: Javaria Ahmed: "RE: Regarding paratool, assign atom types"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
Javaria,
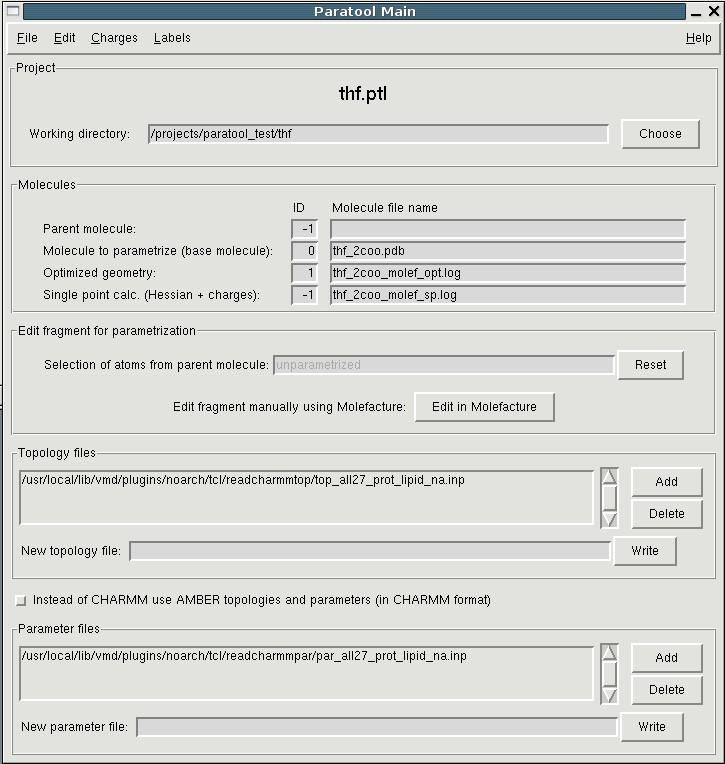
doesn't your paratool main window look like this?:
http://www.ks.uiuc.edu/Research/vmd/plugins/paratool/paratool_main.png
Inthe lower half you can specify the topology files.
Jan
Javaria Ahmed schrieb:
> Hi
>
> Jan, Paratool main screen is not listing any defaultly uploaded topology
> file.
> How can I check , why is that? and how to correct it?
>
> Best Regards,
> Javaria
>
> -----Original Message-----
> From: Jan Saam [mailto:jan.saam_at_charite.de]
> Sent: Mon 1/29/2007 5:56 PM
> To: Javaria Ahmed
> Cc: vmd-l_at_ks.uiuc.edu
> Subject: Re: Regarding paratool, assign atom types
>
> Hi Javaria,
>
> you get the charmm types only if you have loaded the corresponding CHARMM
> topology and parameter files that contain the VDW parameters for Zn and O.
> Paratool should actually load a default CHARMM topology file which
> contains the parameters for ZN and O. The file should be listed in
> Paratool's main screen.
>
> Jan
>
> ---------------------------
> Jan Saam
> Institute of Biochemistry
> Charite Berlin
> Monbijoustr. 2
> 10117 Berlin
> Germany
>
> +49 30 450-528-446
> saam_at_charite.de
>
>
>> Hi
>>
>> I am parameterizing a ZnO molecule using Paratool and following the steps
>> mentioned in Paratool documentation. While assigning atom types, when I
>> proceed to - Choose Type - step, I only get to choose atom types from
>>
> UFF.
>
>> I want to choose atom types Zinc Ion - ZN and Ester Oxygen - OS from
>> charmm
>> force fields instead of choosing some atom type from UFF.
>>
>> How do I get those atom types and their parameters in selection?
>>
>> Regards,
>> Javaria
>>
>>
>
>
>
>
>
>
>
>
-- --------------------------- Jan Saam Institute of Biochemistry Charite Berlin Monbijoustr. 2 10117 Berlin Germany +49 30 450-528-446 saam_at_charite.de
- Next message: ivana adamovic: "Re: About CHARMM electrostaic potential map"
- Previous message: Jan Saam: "Re: Regarding paratool"
- In reply to: Javaria Ahmed: "RE: Regarding paratool, assign atom types"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]



{kind=link}