VMD-L Mailing List
From: Madhur Aggarwal (madhur.aggarwal_at_research.iiit.ac.in)
Date: Wed Oct 02 2019 - 13:02:23 CDT
- Next message: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Previous message: Madhur Aggarwal: "Re: lammps data file simulation box center and VMD origin"
- In reply to: Madhur Aggarwal: "Re: lammps data file simulation box center and VMD origin"
- Next in thread: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Reply: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]
I am extremely sorry I didn't complete my question in my previous mail before hitting enter by mistake ..
So, for visualization, I can use -center origin flag to see my system centred inside one simulation box, but in reality when I run my simulation, as half of my system is outside the box, the pbc conditions will wrap the atoms and the environment for those atoms would change. I do not want this to happen. I want my system should remain well inside one box during the entire simulation (I will also restrict the COM motion to make sure of that).
Because I am specifying my simulation box to be centered at origin itself from the start (by specifying xlo, xhi as -10.0, 10.0 respectively, same for all 3 dimensions) and still it is not working, I am not sure how to achieve this.
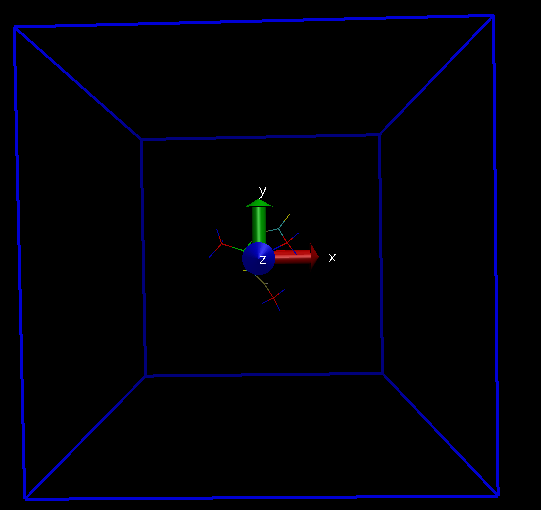
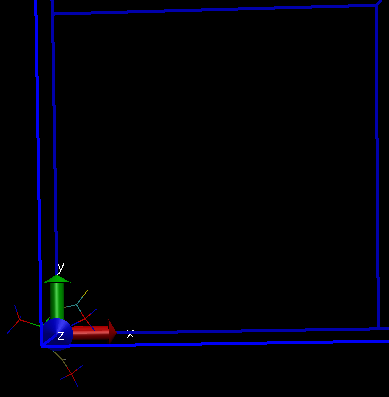
I am attaching a screenshot of my system with simulation box which I get after pbc box command, with and without using -center origin flag.
Thanks again,
Madhur Aggarwal
________________________________
From: Madhur Aggarwal <madhur.aggarwal_at_research.iiit.ac.in>
Sent: Wednesday, October 2, 2019 11:21 PM
To: Bassam Haddad <bhaddad_at_pdx.edu>; Giacomo Fiorin <giacomo.fiorin_at_gmail.com>
Cc: vmd-l_at_ks.uiuc.edu <vmd-l_at_ks.uiuc.edu>
Subject: Re: vmd-l: lammps data file simulation box center and VMD origin
Hi Bassam, Giacomo
Thanks a lot for your prompt reply. I could center my system at the origin now. With the use of "pbc box -center origin" command, I could also center my simulation box at the origin (same as my system). But I have a small doubt here: When I use just "pbc box" command, my simulation box is placed somewhere else (even when I am specifying in my lammps data file to center it on origin (0,0,0)). In this case, onl y a small portion of my system is inside the box and the rest is outside.
So, for visualization, I can use -center origin flag to see my system centred inside one simulation box, but in reality when I run my simulation, as half of my system is outside the box, the pbc conditions will wrap the atoms and the environment for those atoms would change.
________________________________
From: Bassam Haddad <bhaddad_at_pdx.edu>
Sent: Wednesday, October 2, 2019 9:48 PM
To: Madhur Aggarwal <madhur.aggarwal_at_research.iiit.ac.in>
Cc: vmd-l_at_ks.uiuc.edu <vmd-l_at_ks.uiuc.edu>
Subject: Re: vmd-l: lammps data file simulation box center and VMD origin
Hi Madhur,
# select atoms in your system
set all [atomselect top all]
# find the geometric center of the atoms
measure center $all
> x y z (assume these values are not 0)
# move your system to the center (0,0,0)
$all moveby {-x -y -z}
That should do it.
Bassam
On Wed, Oct 2, 2019 at 8:55 AM Madhur Aggarwal <madhur.aggarwal_at_research.iiit.ac.in<mailto:madhur.aggarwal_at_research.iiit.ac.in>> wrote:
Hi all,
I am beginner in VMD. I have a lammps data file which contains the dimensions of my simulation box. I want to translate my system to the center of the box. It would be great if someone could help me in figuring out how to do it.
Also, in my lammps data file, my simulation box dimensions are:
-10.000 10.000 xlo xhi
-10.000 10.000 ylo yhi
-10.000 10.000 zlo zhi
So VMD origin should be at the center of my simulation box, which is not the case when I visualize it by using "pbc box" command. I am not sure why this is happening.
Thanks in advance,
Madhur Aggarwal
- Next message: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Previous message: Madhur Aggarwal: "Re: lammps data file simulation box center and VMD origin"
- In reply to: Madhur Aggarwal: "Re: lammps data file simulation box center and VMD origin"
- Next in thread: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Reply: Axel Kohlmeyer: "Re: lammps data file simulation box center and VMD origin"
- Messages sorted by: [ date ] [ thread ] [ subject ] [ author ] [ attachment ]


