The ability of proteins to fold into their native state is essential for cell function; misfolded proteins not only lose their function, but can also cause neurodegenerative diseases, including Alzheimer and Huntington. Study of protein folding can aid in preventing protein misfolding diseases and in designing proteins with novel functions. Although most cellular proteins fold on timescales of milliseconds to seconds, several small proteins have been designed and characterized experimentally to fold on a timescale of less than 20 µs. The project will employ Center-developed long-time MD simulation with NAMD to investigate: (1) the folding pathway of λ-repressor and (2) the effect of mutations on folding of the λ-repressor.
Spotlight: Protein Hairpin and Parkinson's Disease (Jan 2016)
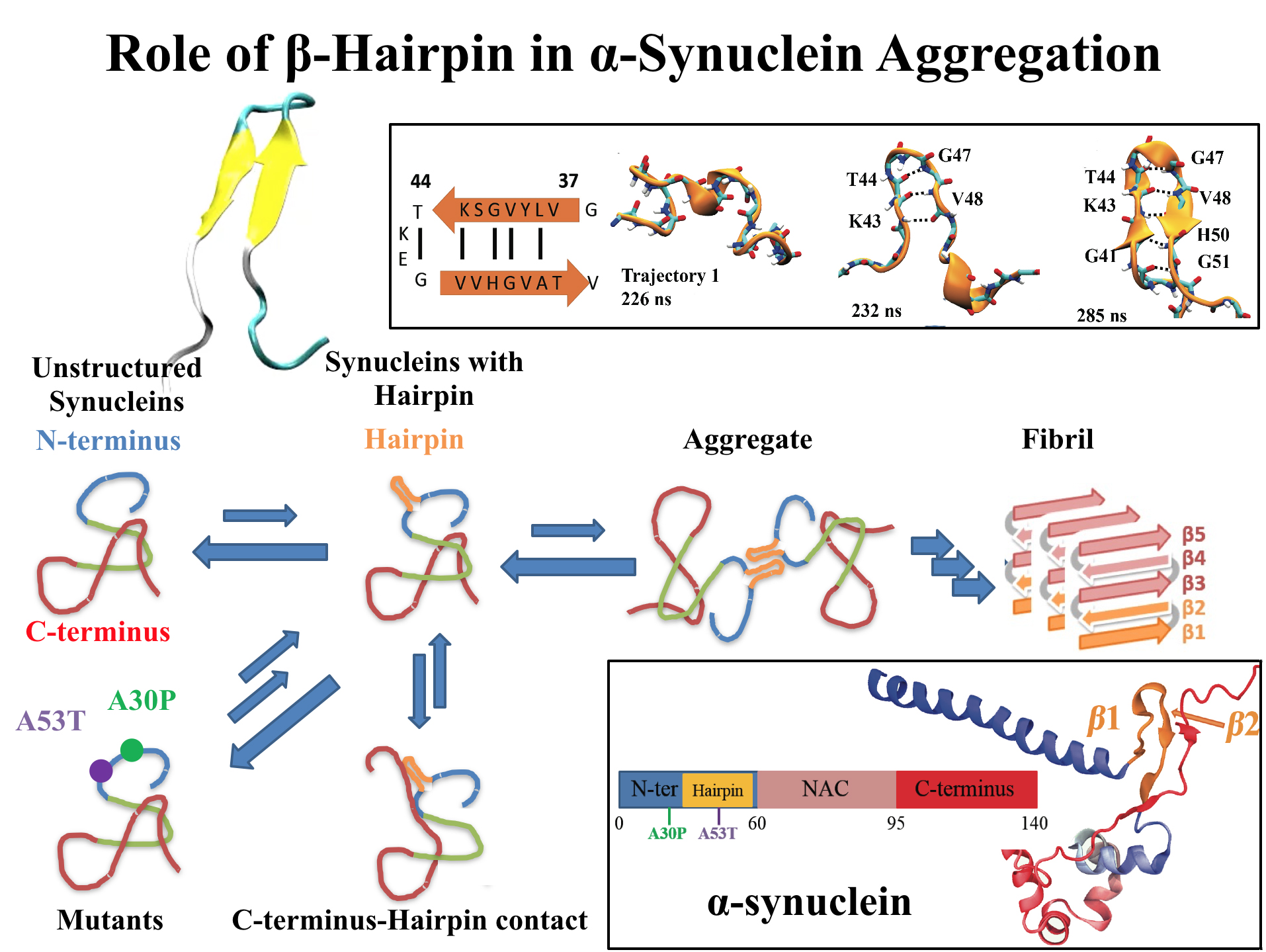
For centuries, millions of people around the globe have been troubled with a movement disorder that usually starts with a tremor in one hand. The disorder, later known as Parkinson's disease, affects commonly older individuals and disrupts patient's movement, sleep and speech from the brain. There is currently no cure for the disease. Key to the disease, progressively occurring in patient's brain, is the loss of neuron cells due to aggregation of a small protein named α-synuclein. Extensive studies have been carried out, yet the function of the protein remains a mystery. It is amazing that aggregation of such small proteins eventually leads to neuronal cell death and generates tremendous difficulties in peoples' life.
In a recent report, a team of computational scientists attributed the cause of α-synuclein aggregation to a hairpin structure involving just a small region (amino acids 38-53) in the middle of the protein. With extensive simulations (over 180 μs in total), the researchers revealed that a short fragment encompassing region 38-53, exhibiting a high probability of forming a β-hairpin structure, is a key region during α-synuclein aggregation. Moreover, the researchers predicted a mutation that impedes β-hairpin formation, thereby retarding α-synuclein aggregation. The discoveries, made possible through the software
NAMD and VMD, are expected to shed light on the mechanism underlying Parkinson's disease and to inspire the design of drugs. More on our α-synuclein website.