Molecular Mechanisms of Retrovirus Infection
Topics: Introduction - HIV mature capsid - RSV immature lattice - Publications - Additional resources - Investigators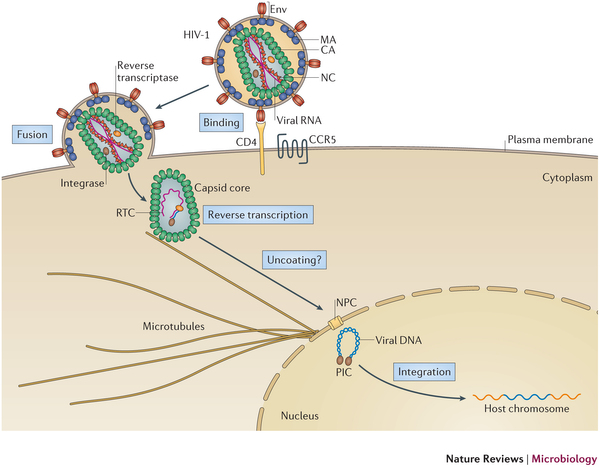
Introduction to retroviruses
The family of Retroviruses are characterized by their ability to incorporate viral DNA into a host cell's genome. Most retroviruses like Rous sarcoma virus (RSV), infect dividing cells during mitosis, when the chromatin is exposed to the cytoplasm. Conversely, the genus of Lentiviruses, like the human immunodeficiency virus (HIV), have evolved in order to infect non-dividing cells. Since the host cell's chromatin is protected by the nucleus, the HIV infection process requires coordination between reverse transcription of viral RNA and nuclear import. Viral RNA is encased in a shell of the capsid protein CA.
Molecular Mechanisms of Human Immunodeficiency Virus Infection
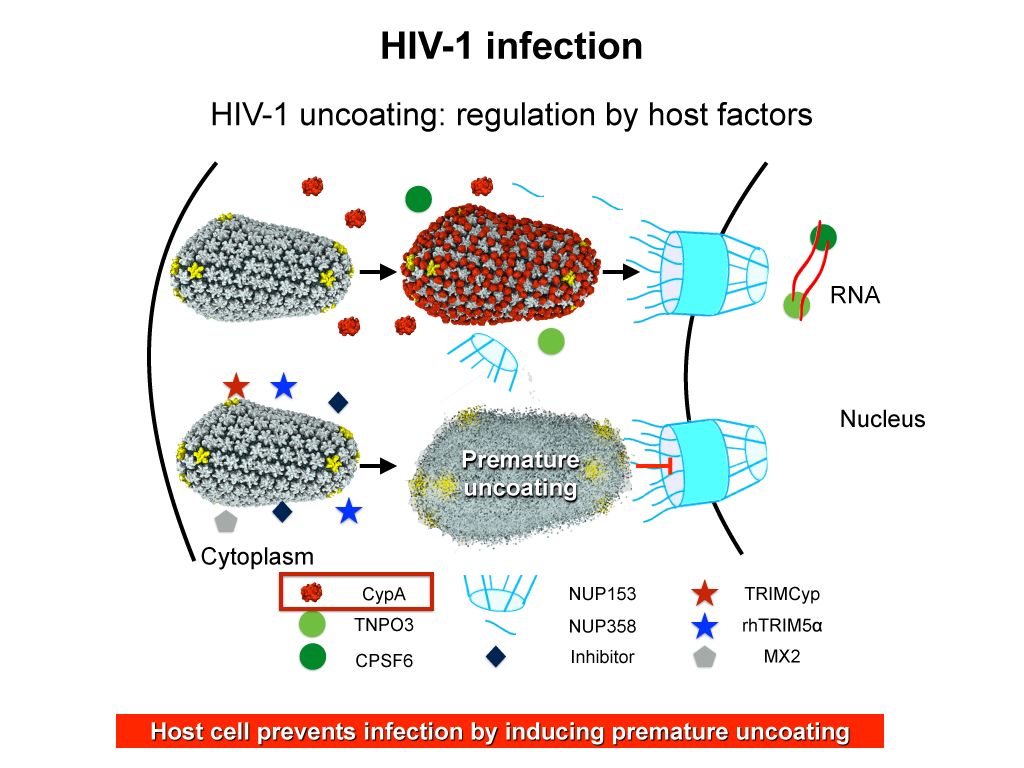
AIDS is caused by the human immunodeficiency virus (HIV). The virus has the ability to infect non-dividing cells, which means that it first needs to establish a pathway into the cell and then permeate the cell's nucleus. For this purpose it enters a cell and recruits cell factors to assist the orchestration of a complex process leading to insertion of the viral RNA into the cell's genome. The infection process involves the capsid surface, a protein shell that encases the viral genome; we just don't know how this process happens. The capsid is a natural therapeutical target as ape cells prevent infection by targeting the capsid. In an experimental-computational collaboration we have successfully established the atomic-level structure of the HIV capsid and could characterize the capsid surface properties, capsid surface processes as well as capsid surface interactions with host proteins and small molecules.
Originally thought to play a trivial role in the infection process, the HIV mature capsid is now well established that the viral capsid fulfills several essential functions. In particular, capsid involvement in the prevention of innate sensor triggering, regulation of reverse transcription, and regulation of the nuclear import pathway is of central importance to the successful infection of a host cell.
Structure of Immature Rous Sarcoma Virus
After a retrovirus is assembled and budded off from the infected host cell, the virus is in its immature form where the viral proteins are arranged in a roughly spherical capsid. These viral proteins, also known as Gag polyproteins, contain the matrix (MA), capsid (CA) and nucleocapsid (NC) proteins. Upon maturation by proteolytic cleavage of Gag, immature retroviruses turn into infectious, mature virions. For RSV and HIV, one of the early cleavage events involves space peptide (SP), situated in between CA and NC. There is currently no consensus on the high resolution structure of SP in its assembled state. Therefore, obtaining an atomic model of immature retroviral lattice that includes the SP domain is paramount to provide valuable structural insight.
An all-atom model of an immature RSV lattice was constructed based on a subnanometer resolution structure of Mason-Pfizer monkey virus (M-PMV) previously obtained via cryo-electron microscopy (cryo-EM). Additional Gag components, namely p10 and SP-NC, were incorporated into the immature lattice model, without which the immature lattice would be unstable. SP-NC was modeled as a six-helix bundle and was simulated for almost 10 µs of replica exchange molecular dynamics simulations. The resulting model of the RSV Gag lattice shows features and dynamics of the capsid protein with implications for the maturation process.
SP is a drug target of great interest because inhibiting the cleavage of SP-NC would severely jeopardize the maturation process of the virus. Preclinical drugs like Bevirimat (BVM) target the SP domain to inhibit the SP-NC cleavage, however they did not pass the clinical trial. Having characterized the structure and the dynamics of an assembled SP would hopefully shed light to the development of next-generation of maturation inhibitors.
Publications
All-Atom Molecular Dynamics of Virus Capsids as Drug Targets
Juan R. Perilla, Jodi A. Hadden, Boon Chong Goh, Christopher Mayne and Klaus Schulten
Journal of Physical Chemistry Letters, 2016, in press
Contributions of charged residues in structurally dynamic capsid
surface loops to Rous sarcoma virus assembly.
Katrina
J. Heyrana, Boon Chong Goh, Juan R. Perilla, Tam-Linh N. Nguyen,
Matthew R. England, Maria C. Bewley, Klaus Schulten and Rebecca
C. Craven
Journal of Virology, 2016, in press
Cyclophilin A stabilizes the HIV-1 capsid through a novel non-canonical binding site.
Chuang Liu*,Juan R. Perilla*, Jiying Ning, Manman Lu, Guangjin Hou, Ruben Ramalho, Benjamin A. Himes, Gongpu Zhao, Gregory J. Bedwell, In-Ja Byeon, Jinwoo Ahn, Angela M. Gronenborn, Peter E. Prevelige, Itay Rousso, Christopher Aiken, Tatyana Polenova, Klaus Schulten and Peijun Zhang
Nature Communications , 2016, 10.1038/ncomms10714
Molecular
architecture of the retroviral capsid.
Juan R. Perilla and Angela
M. Gronenborn
Trends in Biochemical Sciences, 2016, in press
Dynamic allostery governs cyclophylin A-HIV capsid interplay.
Manman Lu, Guangjin Hou, Huilan Zhang, Christopher L. Suiter, Jinwoo Ahn, In-Ja L. Byeon, Juan R. Perilla, Christopher J. Langmead, Ivan Hung, Peter L. Gor'kov, Zhehong Gan, William Brey, Christopher Aiken, Peijun Zhang, Klaus Schulten, Angela M. Gronenborn, and Tatyana Polenova.
Proceedings of the National Academy of Sciences USA,2015, 47,14617-14622
Chemical visualization of human pathogens : the Retroviral Capsids.
Juan R. Perilla*, Boon Chong Goh*, John Stone, and Klaus Schulten. Proceedings of the 2015 ACM/IEEE Conference on Supercomputing, 2015
Atomic Modeling of an Immature Retroviral Lattice Using Molecular Dynamics and Mutagenesis
Boon Chong Goh*, Juan R. Perilla*, Matthew R. England, Katrina J. Heyrana, Rebecca C. Craven, and Klaus Schulten, Structure, 23:1-12, 2015
Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics.
Gongpu Zhao* , Juan R. Perilla*, Ernest L. Yufenyuy*, Xin Meng, Bo Chen, Jiying Ning, Jinwoo Ahn, Angela Gronenborn, Klaus Schulten, Christopher Aiken, and Peijun Zhang. Nature, 497:643-646, 2013.
Additional resources
MoviesFigures
PDB structures
We published two representative structures of the HIV-1 capsid structure. To load these structures into VMD please use the following script:For 3J3Y:
foreach i {1VUU 1VUV 1VUW 1VUX 1VUY 1VUZ 1VV0 1VV1 1VV2 1VV3 1VV4 1VV5 1VV6 1VV7 1VV8 1VV9 1VVA 1VVB 1VVF 1VVG 1VVH 1VVI } { mol new $i }
For 3J3Q :
foreach i { 1VU4 1VU5 1VU6 1VU7 1VU8 1VU9 1VUA 1VUC 1VUD 1VUE 1VUF 1VUG 1VUH 1VUI 1VUJ 1VUK 1VUL 1VUM 1VUN 1VUO 1VUP 1VUQ 1VUR 1VUS 1VUT } { mol new $i }
RSV: The homology model of the immature RSV lattice reported in (Structure, 23:1-12, 2015) is available here for download.
Molecular dynamics trajectory of capsid structure 3J3Y.
In (Nature, 497:643-646, 2013) we report a 64 million atom molecular dynamics simulation of structure 3J3Y that revealed stability as seen in a 100 ns trajectory. This claim is corroborated here through snapshots of the trajectory at 50 ns, 60 ns, ... 100 ns to which we add for the sake of comparison the pdb entry 3J3Y. The snapshots can be viewed through VMD.
To open the trajectory simply use VMD to open the PSF file (File -> New Molecule -> HIVcapsid.pdf), and then add the dcd file (File -> Load Data Into Molecule -> HIVcapsid.dcd).
The reader can inspect that the simulated structure indeed remains stable and close to the structure reported in the protein data base. Further details of the trajectory will be reported in a forthcoming publication.
In releasing the trajectory data we follow the example of David Shaw Research who likewise released trajectory data for protein folding, except that we do not require registration and signing of a license. The latter generosity is simply dictated by our lack of legal support, but we expect that the use of the trajectories is properly acknowledged through explicit reference of use of the trajectories and of Nature, 497:643-646, 2013.
Readers who wish to carry out their own molecular dynamics simulations are referred to the VMD website, in particular to the mini-tutorial for solvating large structures, as well as to the NAMD web site, in particular to instructions to run NAMD for very large structures. The program NAMD is described here and its application in a simulation of a very large (100 Million atom) system based on NAMD 2.8 is described here.
Investigators
- Juan R. Perilla
- Boon Chong Goh
- Christopher Aiken
- Rebecca Craven
- Angela Gronenborn
- Tatyana Polenova
- Peijun Zhang
- Gongpu Zhao
Page created and maintained by Juan R. Perilla and Boon Chong Goh.


