Highlights of our Work
2026 | 2025 | 2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
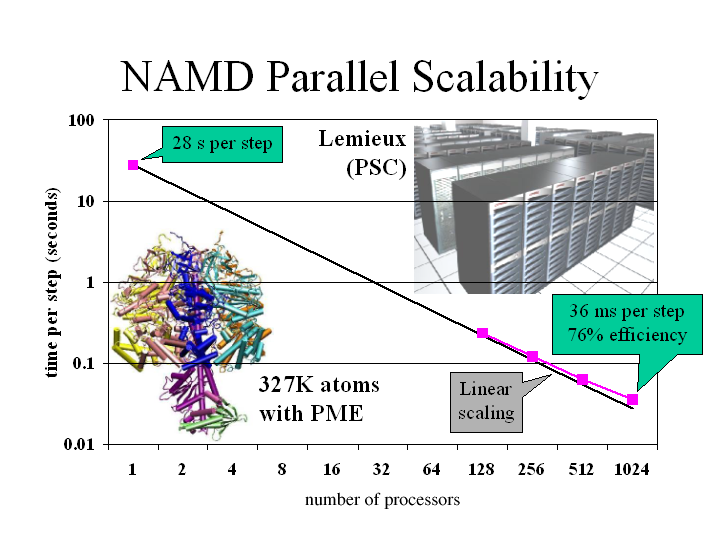
image size: 150.4KB
The parallel molecular dynamics program NAMD, and its sister visualization program VMD, have helped researchers at Illinois discern how muscles stretch, nerves sense pressure, and kidneys filter water. The latter project, for example, used simulations of 106,000 atoms to discover how aquaporins, which are ubiquitous in mammals, plants, and bacteria, allow water to pass while preventing the conduction of protons or ions (article in Science, more). Our years of work developing this software to apply the nation's fastest sup ercomputers to understand the tiny components of living cells were recognized at the SC2002 High Performance Networking and Computing conference with a Gordo n Bell Award for unprecedented parallel performance on a challenging computational problem (pdf of paper-497k). NAMD and VMD are distributed , free of charge, to thousands of scientists in industry and academia around the world, quickening the pace of drug discovery and other vital research to unravel biological processes.
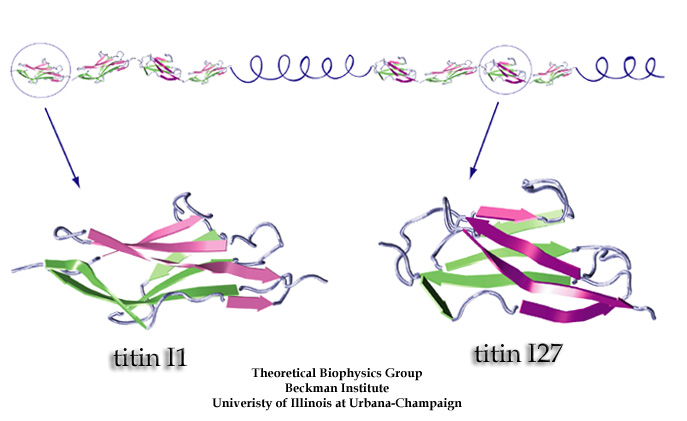
image size:
71.7KB
made with VMD
What kind of function does the longest gene in the human genome code for? The answer is a bit mundane: a very long molecular spring that provides muscle with passive elasticity. Nature adjusts the protein, called titin, for many types of muscle, e.g., skeletal or cardiac muscle, as well as for other cellular functions. The molecular spring contains hundreds of elastic elements in series like beads on a string. One type of bead is the immunoglobulin domain, which can stretch to ten times its normal length. For a long time only one of the immunoglobulin domains was structurally known, permitting only a single peek into nature's design library. Recently, a second domain became structurally known and protein crystallographers and modelers joined forces to discover how nature designs its beads along titin, as described in a recent publication.
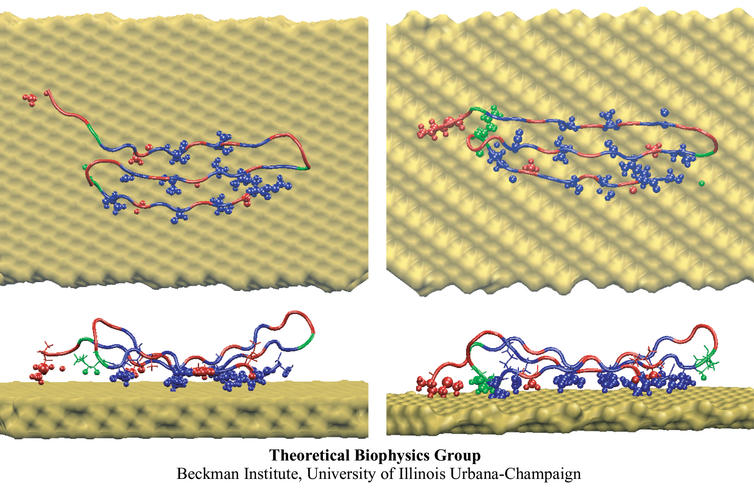
image size:
91.6KB
made with VMD
The biological control of inorganic crystal formation, morphology and assembly is of interest to biologists and biotechnologists studying hard tissue growth and regeneration, as well as to materials scientists using biomimetic approaches for control of inorganic material fabrication and assembly. A molecular-level understanding of the natural mechanisms involved in these processes can be derived from the use of metal surfaces to study surface recognition by proteins together with combinatorial genetics techniques for selection of suitable peptides.
In a recent study, the structure of a genetically engineered gold binding protein has been determined computationally, and the ability of the protein to recognize gold crystal surfaces has been explained. Molecular dynamics simulations were carried out with the program NAMD using the solvated protein at the gold surface to assess the dynamics of the binding process and the effects of surface topography on the specificity of protein binding.
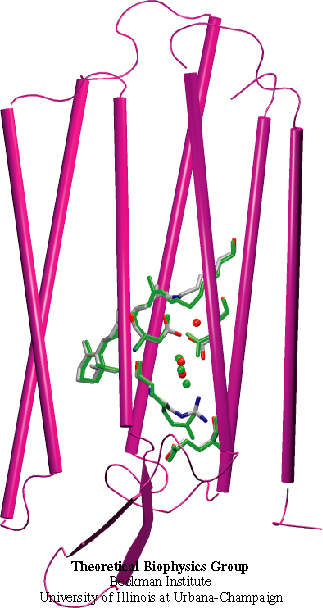
image size:
139.9KB
made with VMD
Resolving the physical processes that underly the biological function of a protein can be an elusive goal even with extremely detailed observations available. An example is the protein bacteriorhodopsin, a light driven proton pump in archaebacteria. This protein is a close relative to human G-protein coupled receptors that are the target for many pharmacological interventions and, hence, knowledge of bacteriorhodopsin's dynamics is of great medical interest. Despite the availability of highly resolved structures and spectroscopic observations of the protein and its functional intermediates, as they arise within 10-12 s of light absorption triggering its function, the physical mechanism remained ill understood. A recent computational modeling study that combined a quantum mechanical simulation of the protein's active site with a classical mechanical simulation of the remainder of the protein succeeded to fill in the elusive detail that reveals a complete picture of how the protein initiates proton pumping, a key step to explain entirely the biological function. For more information see here.
image size:
62.3KB
made with VMD
Light is fundamental for life. Through many photosynthetic life forms, its energy fuels the major part of Earth's biosphere. The familiar green color of plants, so ubiquitous in our surroundings, stems from chlorophylls, molecules that help plants, algae, and some bacteria to harvest the sunlight. Recently, the structure of an apparatus that harvests sunlight in cyanobacteria, and actually in a similar fashion in plants, has been discovered, showing 96 chlorophylls being held at close distances by a protein complex. The chlorophylls absorb sunlight and deliver its energy to a central chlorophyll pair that utilizes it to electronically charge a cell membrane, the whole functioning like an extremely efficient biological solar cell. Quantum physics and a theoretical analysis of the energy utilization of the system, reported in a recent publication, have revealed that this system has been designed with a high degree of fault tolerance and optimality: pruning single and even multiple chlorophylls hardly affects the efficiency of the apparatus; altering the chlorophylls' arrangement though leads to a reduction of efficiency. Since the apparatus is naturally exposed to intense radiation and subject to continuous damage, its robustness is crucial for the organism.
image size: 379.0KB
The building blocks of living cells are biomolecules so small that no light microscope can see them, yet viewing them is essential to decipher the inner workings of cells. The best looking-glass for biomolecules (such as proteins) available today is computers running molecular graphics software that translates experimental data into the molecular graphics. Now the wide availability of molecular graphics has taken a step forward with our new visualization package, JMV (Java Molecular Viewer). JMV borrows several key features from our visualization tool for large scale biomolecules, VMD. The JMV applet places the picture of a protein in your web browser, shown in a 3-D view, ready to be rotated, scaled, and colored according to physical properties. JMV will serve the next generation of bioinformatics web tools, like BioCoRE, through its great adaptability and will turn every molecular picture in electronic text books or web sites into an interactive looking-glass.
image size:
104.1KB
made with VMD
Living cells rely on nutrients absorbed through their cell membranes, for example on glycerol that is key to the cells' metabolism. Proteins, so-called aquaporins, in the membranes form channels that act as sieves permitting passage of water, glycerol, and like molecules, but prevent other molecules of similar size from entry and clogging. For this purpose the channels interact strongly with molecules attempting to pass. In a recent publication, the energetics of the conduction process of glycerol for the aquaporin GlpF was measured in a molecular dynamics simulation, carried out with NAMD, that pulled glycerol through the channel, monitoring the forces needed to advance along the channel axis. An analysis that discounted the irreversible work done on glycerol, a difficult prerequisite, yielded the energy profile that glycerol experiences along the channel and that reflects how the protein decides which molecules are allowed to pass the sieve.
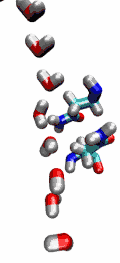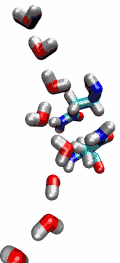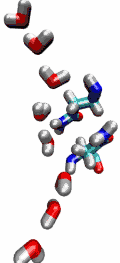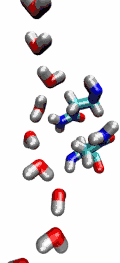
image size:
82.5KB
made with VMD
Human kidneys need to filter about a bathtub of water a day through cells that contain membrane channels made of proteins called aquaporins. Crystallographers from the University of California at San Francisco (R. Stroud and coworkers) that discovered the structure of one type of aquaporins, aquaglyceroporins, have teamed up with UIUC researchers to determine how these channels achieve their very high water throughput, yet prevent the cells' electrical potential from discharging by not permitting any flow of ions or conduction of protons. The team, combining 106,000 atom simulations using NAMD and crystallography, found that the channels achieve the impressive filtering function by conducting water single file and keeping the water molecules strictly oriented: water molecules enter the channel oxygen atom first and leave the channel oxygen atom last. Aquaporins are ubiquitous in mammals, plants, and bacteria and the finding, published recently in Science magazine, has implications for many biological functions as well as for human diseases, e.g., cataract of the eye, loss of hearing, or diabetes insipidus. (more, press release)
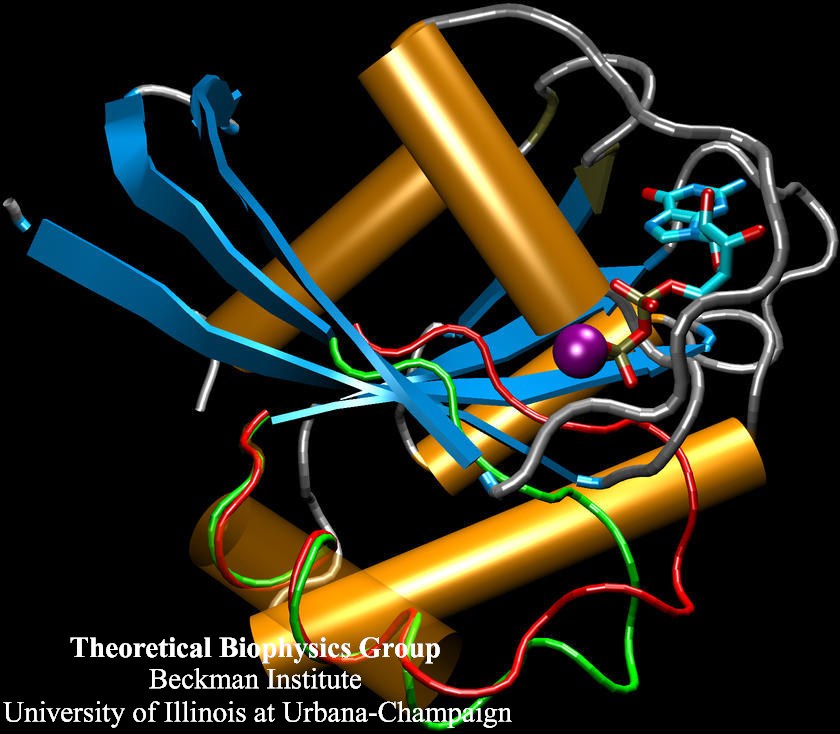
image size:
98.9KB
made with VMD
Biological cells process numerous types of information, for optimal control of their life cycles or to adapt to their environment, and recruit for this purpose signaling proteins. The best known among the latter are the G-proteins, involved in numerous diseases and related to many targets of drugs. G-proteins are closely related to motor proteins: G-proteins get switched on and off through the binding of GTP and its hydrolysis to GDP; motor proteins generate mechanical force through binding of ATP and its hydrolysis to ADP. A recent publication reports a 19,463 atom computer simulation using NAMD that reveals a "power stroke" in G-proteins likewise found in motor proteins. The stroke switches on and off G-proteins' ability to interact with other signaling proteins, with a power stroke that transforms the protein from an ordered into a disordered conformation.
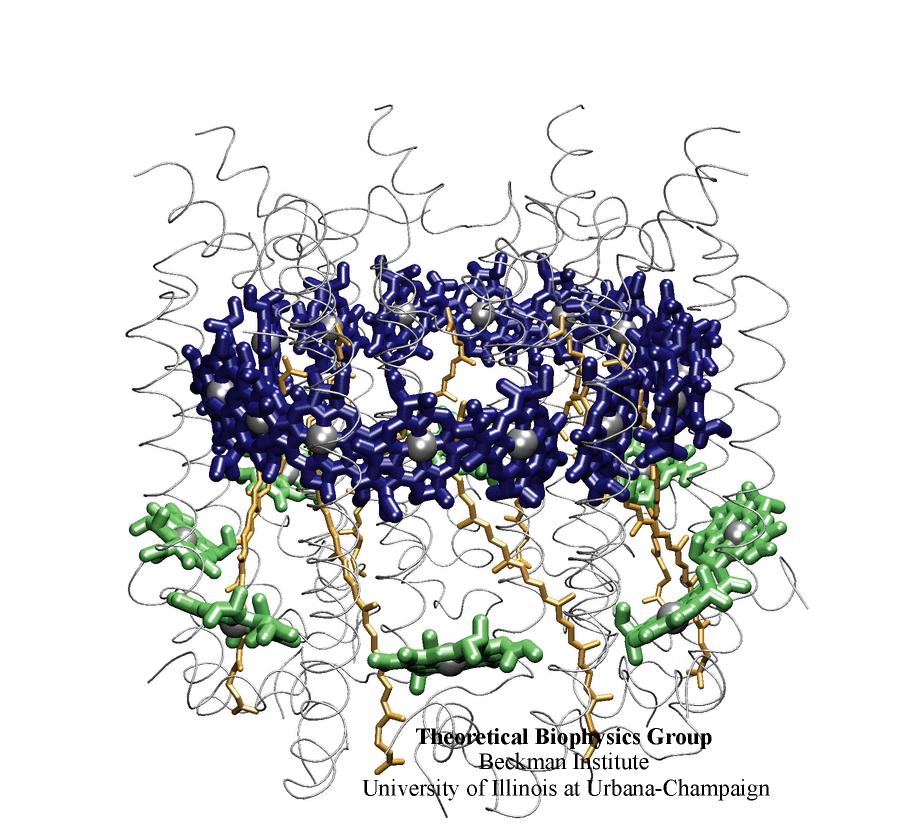
image size:
122.6KB
made with VMD
Most life forms exist near temperatures of about 300 Kelvin where thermal disorder is significant. Understanding how life copes with this disorder, in fact, most often exploits it, poses a deep intellectual challenge. Two recent publications investigate thermal disorder for electronically excited bioelectronic systems that harvest sun light and funnel its energy into the metabolism of so-called purple bacteria. One study borrows mathematics (supersymmetric calculus) from the physics of elementary particles to describe the optical properties of randomly distributed, but otherwise immobile, aggregates of chlorophylls. The second study goes a step further and investigates optical properties affected by thermal motion. The paper draws its insights from a pioneering 87,055 atom molecular dynamics simulation of a membrane-protein-chlorophyll system that monitored thermal motion of atoms and electrons and extends a mathematical description, the polaron model, used in advanced solid state physics.
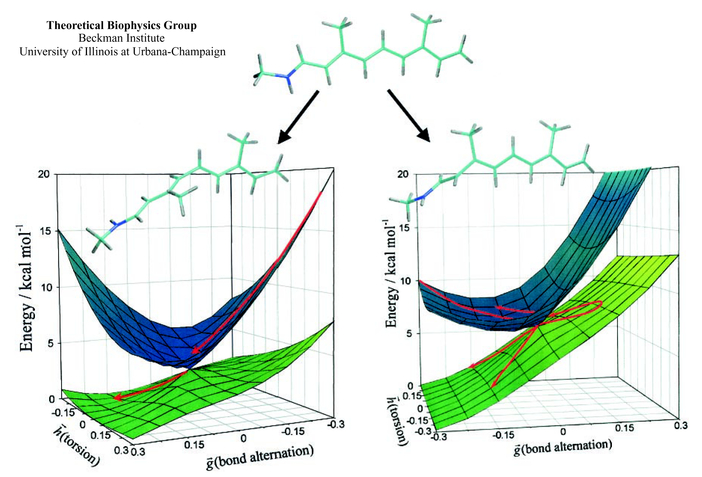
image size: 271.6KB
Since Newton, vision has attracted physicists seeking to explain how light is sensed by organisms. Recently, the structure of a visual receptor protein has been solved crystallographically and physicists have a new opportunity to explain vision in atomic level detail. Vision starts with optical excitation of retinal, located in the receptor protein, and the subsequent vibrational - torsional motion in retinal's electronically excited state. Retinal reaches within less than a picosecond (0.000000000001 s) geometries for which excited state and ground state merge energetically, the so-called conical intersections. Here retinal converts back to the ground state and becomes trapped into a new stable geometry. A recent study by the Theoretical Biophysics Group explains how the conical intersections of retinal steer retinal towards the right trapped geometry, one that is capable of triggering a visual signal.
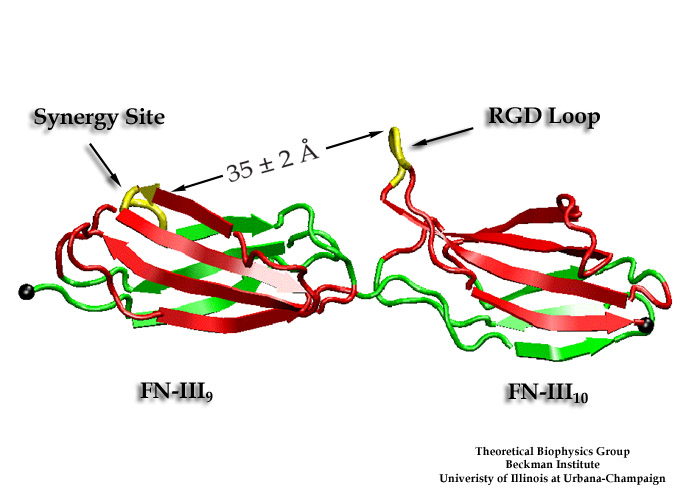
image size:
85.1KB
made with VMD
Cells in animals adhere to dynamic, seemingly random assemblies with other cells that make up tissues like skin, organs, and brain. The cell's adhesion and motion is controlled by the extracellular matrix, with the protein fibronectin being a key component. The proteins have optimal mechanical elasticity and also signal to cell surface receptors, integrins, the tension exerted on them. How this is achieved is the subject of an ongoing collaboration with the research group of Viola Vogel of the Department of Bioengineering at the U. of Washington in Seattle (see also Oct 2001 highlight). The most recent publication from this effort reports a 97,884 atom steered molecular dynamics simulation using NAMD. It is revealed now that stretching two consecutive domains of fibronectin deforms two sites, the so-called RGD and synergy sites as well as their distance. This weakens binding to cell receptors and, as a result, integrins can function as gauges that signal the magnitude of exterior forces to a cell.

image size:
66.6KB
made with VMD
Adenosine triphosphate (ATP) is the fuel of life; every living cell must use ATP to carry out its functions, and the human body synthesizes its own weight in ATP every day. The ubiquitous molecular motor ATP synthase catalyzes the creation of ATP by precisely directing electrochemically generated torque. A detailed understanding of how this system functions can impact areas ranging from neurodegenerative disease research to nanotechnology development. Running at the Pittsburgh Supercomputing Center on LeM ieux, the most powerful computer system in the world for open research, the freely available simulation code NAMD can simulate a solvated all-atom model of ATP synthase with full electrostatics at 65% efficiency on 1000 processors. This achievement in scalability places NAMD an order of magnitude ahead of comparable packages for molecular dynamics simulation.
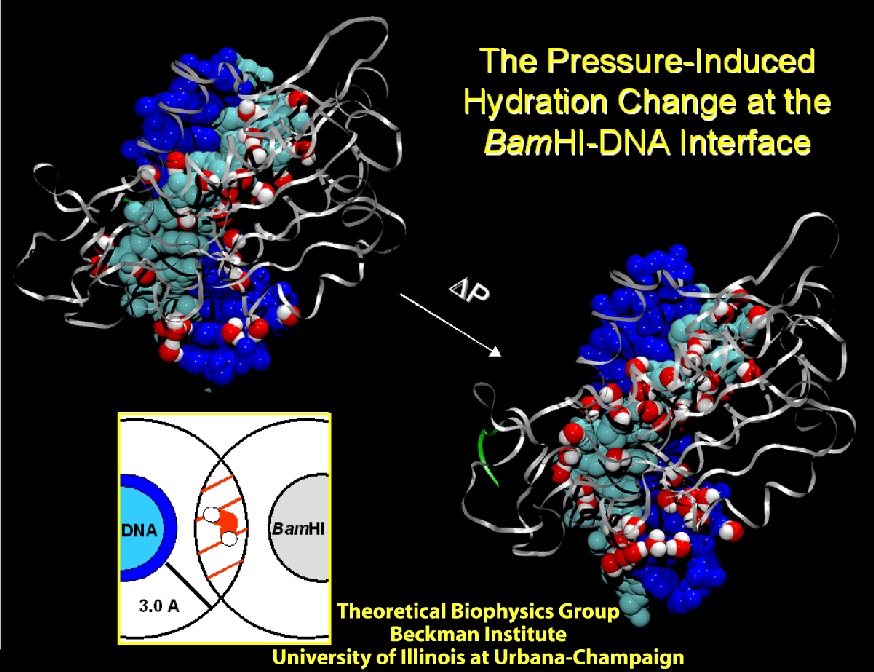
image size:
225.3KB
made with VMD
Deciphering the processes by which proteins recognize and bind to DNA is critical in our quest to understand cellular functions. To reach this goal, a collaboration with the group of Stephen Sligar, UIUC, explored the factors involved in protein-DNA recognition using hydrostatic pressure to perturb the binding of the BamHI endonuclease to cognate DNA. Our joint resulting publication outlines a new technique of high-pressure gel mobility shift analysis to test the effects of elevated hydrostatic pressure on the binding of BamHI (so-called restriction enzyme) to a specific DNA sequence. Upon application of a hydrostatic pressure of 500 bar, recognition between BamHI and the DNA sequence was weakened nearly 10-fold, suggesting an important role of water. An advanced 65,000 atom nanosecond molecular dynamics simulations with NAMD, at both ambient and elevated pressures, complemented the experiments and revealed how water-mediated interactions between BamHI and DNA control sequence recognition.


