Highlights of our Work
2024 | 2023 | 2022 | 2021 | 2020 | 2019 | 2018 | 2017 | 2016 | 2015 | 2014 | 2013 | 2012 | 2011 | 2010 | 2009 | 2008 | 2007 | 2006 | 2005 | 2004 | 2003 | 2002 | 2001
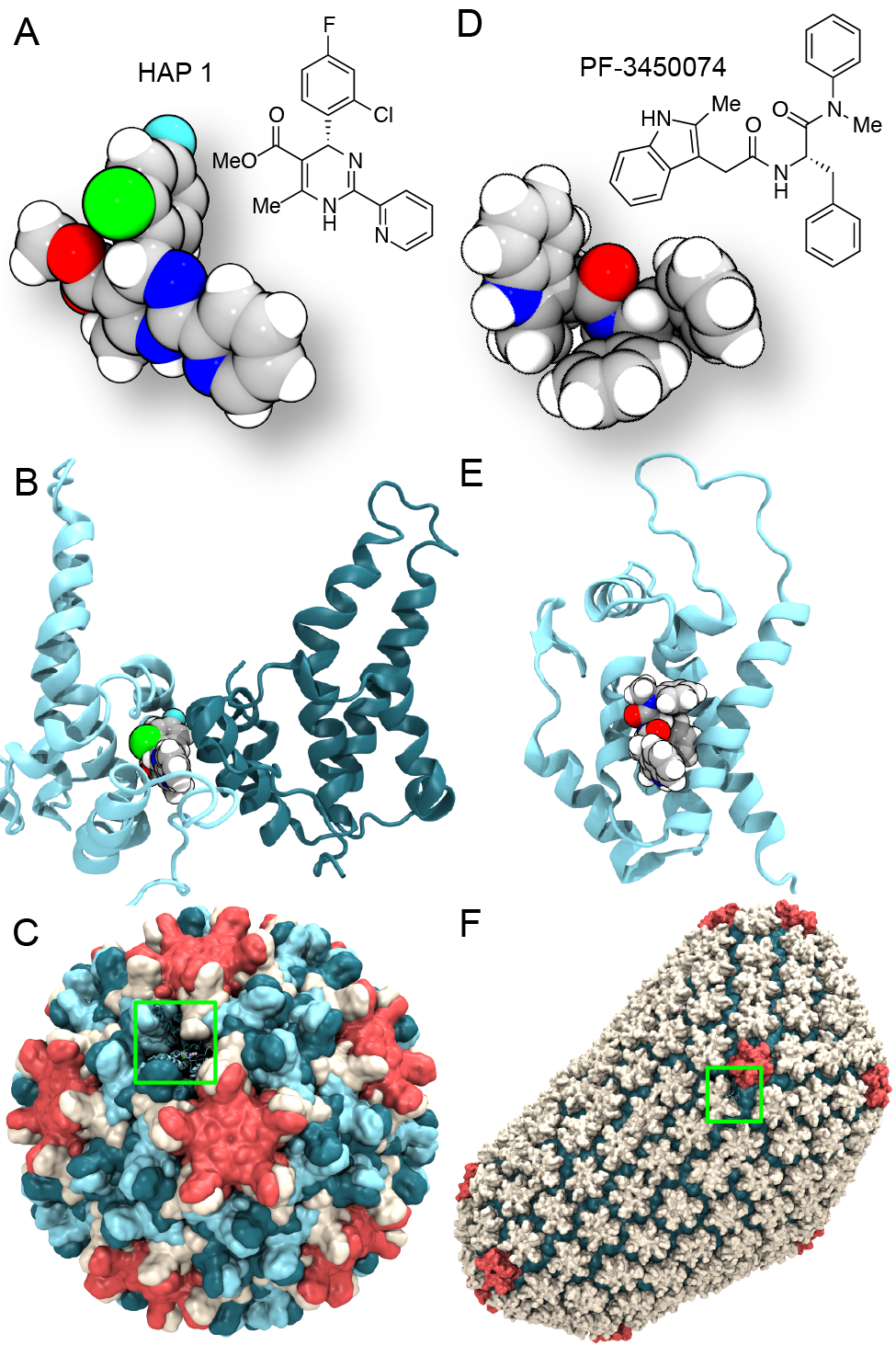
image size:
0 bytes
made with VMD
Virus capsids, specialized protein shells that encase the genome of
viral pathogens, play critical roles in regulating viral infection,
and are, thus, of great pharmacological interest as drug targets. In
particular, small-molecule drugs (typically <900 Da) represent a
promising antiviral strategy, and a number of such compounds have been
developed to inhibit virus capsids by interfering with their assembly
and uncoating processes. Importantly, to explain the mechanisms by
which drugs disrupt capsid function, as well as to successfully design
novel therapeutics, the interactions of drug molecules with their
capsid targets must be studied at full chemical detail. Toward this
end, all-atom molecular dynamics simulations are emerging as an
essential technique to investigate the effects of small-molecule drugs
on capsid structure and dynamics. Research presented in a recent Perspective
applying simulations in NAMD
to study drug-bound hepatitis B virus (HBV) and
human immunodeficiency virus type 1 (HIV-1) capsids suggests the types
of valuable chemical and physical information computational approaches
can reveal, and underscores the importance of simulating, not isolated
capsid proteins, but functional assemblies up to the level of complete
capsids.
Notably, through analysis with VMD,
the study found that binding of the drug HAP1 to the HBV capsid
causes global structural changes that subtly alter the overall capsid shape,
including a flattening of capsid curvature. Further, the study found that the
binding of the drug PF74 to the HIV-1 capsid imposes rigidity and causes
shifts in allosteric communication pathways connecting distant regions of the
capsid protein. The authors of the Perspective anticipate that many other such
exciting discoveries regarding virus capsid function and their use as drug
targets lie just ahead on the horizon, and molecular dynamics simulations will
drive these discoveries pending a series of notable advancements in
computational methodology.


