Automatic PSF Generation Plugin, Version 1.8
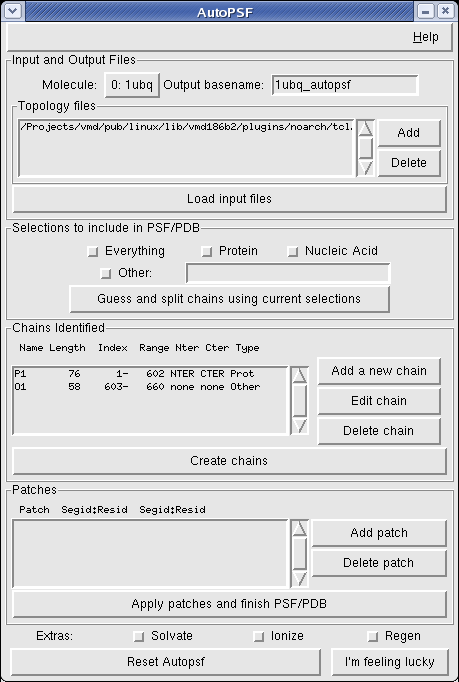
The AutoPSF plugin provides a graphical and text interface for automatically generating a dynamics-ready psf and pdb file from a raw pdb, obtained from the Protein Data Bank or elsewhere. It is designed to provide a fast and intuitive interface for psfgen, the actual backend for PSF generation. While AutoPSF makes educated guesses about most aspects of your structure, it is imperative that you check the sanity of the final result, especially with regards to terminii and the location of chain breaks. In addition, some features of psfgen are not yet available in AutoPSF. This page provides documentation on basic use of AutoPSF. For more detail, please read this step by step technical description of how it works and what it does.
Using the AutoPSF graphical interface
The AutoPSF GUI will walk you through the process of psf generation step by step. Different sections of the GUI will be enabled or disabled as you go through the procedure. Two buttons at the bottom are available at any point: Reset Autopsf (which will return you to the beginning) and I'm feeling lucky (which will attempt to finish the PSF and PDB automatically with the information you have given so far. The stages of normal use are as follows:
Input and Output Files
AutoPSF acts on exactly one currently loaded input pdb file, specified by the Molecule pulldown menu. The output files will have prefixes specified by Output basename. You may specify one or more topology files in the Topology files menu; a sensible default file is provided, but you will need to include others if you have nonstandard residues in your structure.
Selections to include in PSF/PDB
The checkboxes in this section select which parts of your loaded pdb will be included in the final structure. Leaving "Everything" checked will cause the entire pdb to be included in the final structure. Protein and Nucleic Acid components may also be selected. The "Other selections" box allows you to type in an arbitrary VMD selection text for extra atoms to include. This can be useful, for example, to include a ligand molecule but exclude crystallographic waters.
Chains Identified
The "Chains Identified" dialogue shows a list of chains identified in your structure using the selections from the previous section. Each chain includes a length (in residues), start and end atomic indices, and the N and C terminal patches to be applied. If the chain selections it has made are incorrect, you may edit, add, or delete chains from the list. Once you have the proper chains chosen, clicking "Create chains" will parse your input files and find any non-terminal patches which AutoPSF things are needed.
Patches
The "Patches" menu provides an interface for adding or removing patches to be applied to your structure. A number of patches, such as disulfide bonds, may be automatically generated by AutoPSF and will be shown here. New patches may be applied, or displayed patches deleted, using the "Add patch" and "Delete patch" buttons. Once you are finished, clicking "Apply patches and finish PSF/PDB" causes AutoPSF to run psfgen. The finished psf and pdb files will be be named BASENAME.psf and BASENAME.pdb, where BASENAME is the Output basename that you specified. Note that it is necessary to push this button to finish the PDB even if no patches are displayed.Extras
A small number of additional features may be applied at the end of the autopsf run. The "Solvate" and "Ionize" options will run the plugins solvate and autoionize (using sensible default settings) after psf generation is complete. The "Regen" option will cause psfgen to regenerate all angles and dihedrals from connectivities prior to writing the psf. Note that this step is generally not necessary, but is needed with early generations of CMAP-based topology files if your molecule includes any patches.
Fragment chooser
Normally, clicking the Create PSF/PDB button will generate your psf and pdb automatically, but this is not possible if your molecule contains residues that are not specified in your topology file(s). In that case, an extra dialogue box will appear that offers you the choice to delete the fragments from your selection and rerun psfgen, or parameterize the unknown residue using paratool.
Using AutoPSF text commands
AutoPSF can also be run from the Tcl console, in which case it will run straight through the PSF generation procedure without interaction. It can be used by typing autopsf -mol MOLID in the console, where MOLID is the number of the molecule you wish to act on. By default, autopsf will use its internal topology file, and act on the entire molecule. It recognizes the following options:
- -protein: Include protein. Note that if protein or nucleic acid are speficied, all other parts of the molecule will be excluded by default unless they are explicitly specified.
- -nucleic: Include nucleic acid residues. As with protein, if this is specified, all other parts of the molecule will be turned off by default.
- -include seltext: Adds the atoms specified by selection text to the section that will be autogenerated. This is useful in tandem with protein or nucleic acid to select specific cofactors for inclusion without using, for example, crystallographic water. Only one -include statement should be used for a given autopsf run.
- -solvate: Solvate the protein in a water box after psf generation. This option will use default settings for solvate, and pad the solvation box to include 12 angstroms in each direction. If this step is ommitted, the VMD solvate module may be used later for more control.
- -ionize: Include NaCl in sufficient quantity to neutralize the protein and provide an overal ionic concentration of 0.5 M. If this option is omitted, the VMD autoionize module may be used later for more control.
- -prefix string: Sets the prefix for the finished .psf and .pdb files; the default of autopsf will be used if this value is not specified.
- -top: In GUI mode, pushing this button will open a dialog to allow additional topology files to be specified (in their absence, a suitable default will be chosen). In command line mode, topology files are specified at run time using this switch.
- -noguess: Don't guess coordinates of missing atoms (not recommended)
- -noterm: Don't automatically add terminii to proteins and nucleic acid segments (not recommended)
- -gui: Force creation of gui from command line
- -regen: Regenerate angles and dihedrals.
- -patch: Apply a patch. The patch should be enclosed in double quotes, and specified as "PATCHNAME CHAIN1:RES1 CHAIN2:RES2"


