Processes Involved in Infection of Human Cells by the SARS-CoV2 Virus
Peptide Inhibitor Design for Targeting SARS-CoV2 Spike Protein
|
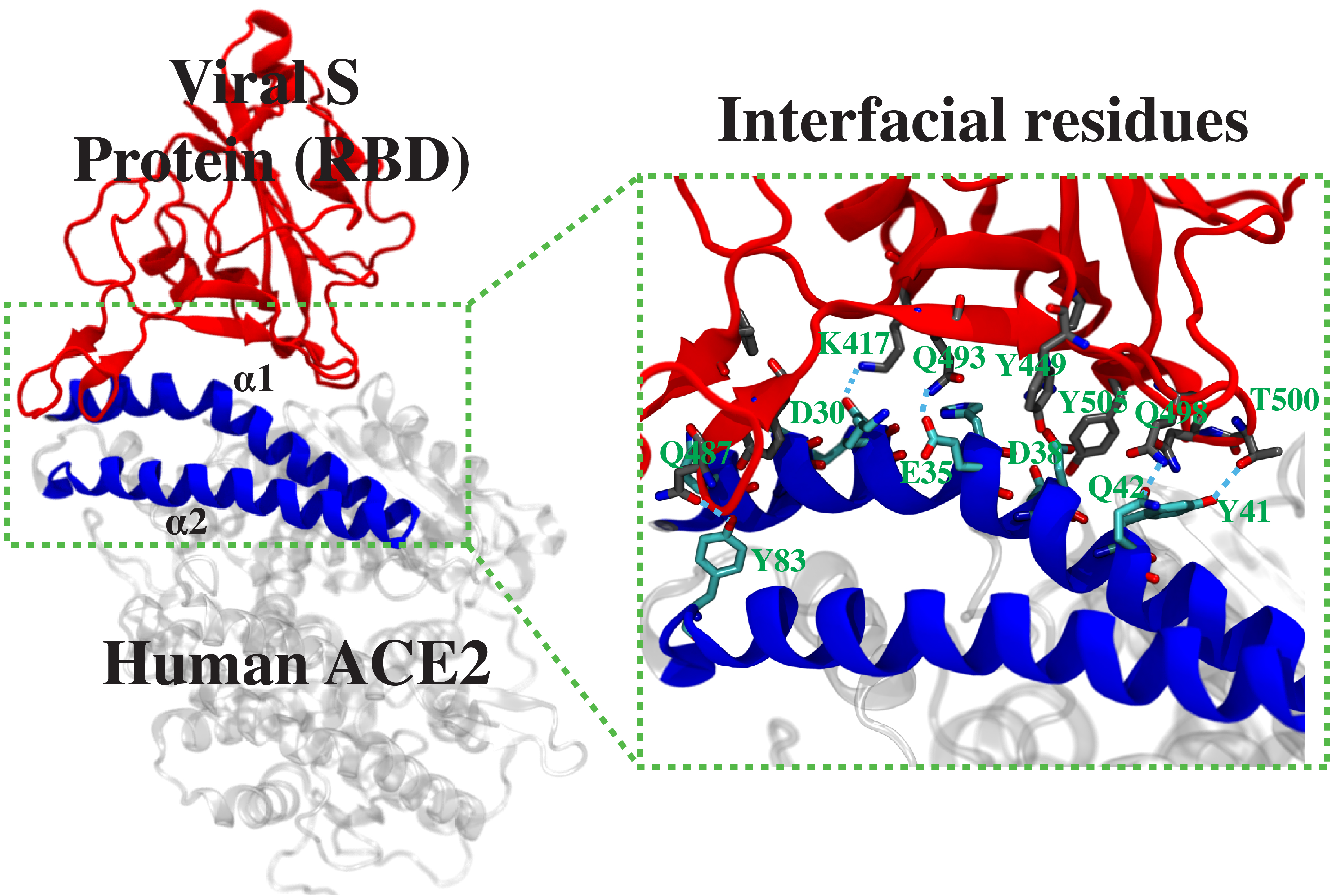
This project is part of a collaboration among several research labs of the University of Illinois at Urbana-Champaign to develop a peptide inhibitor to block the spike protein of SARS-CoV2 from its binding to the human angiotensin-converting enzyme 2 (ACE2). The design originates from a peptide fragment corresponding to the region of ACE2, where the SARS-CoV2 spike protein binds with high affinity. Here we will first use equilibrium MD to examine the stability of the complex between the receptor-binding domain (RBD) of the SARS-CoV2 spike protein and each of the 40 different ACE2-derived peptides. The ten best candidates showing stable complex with the spike-RBD will be further analyzed by well-tempered metadynamics to rigorously calculate their binding free energies, in order to select the best peptide sequence design. Furthermore, using a similar approach, we will develop a second-generation peptide inhibitor with additional modifications to increase the binding affinity to the spike-RBD.
This is an active project under the
COVID-19 HPC Consortium.
|
|
Simulation of SARS-CoV2 Spike Proteins in Crowded Viral Envelope
|
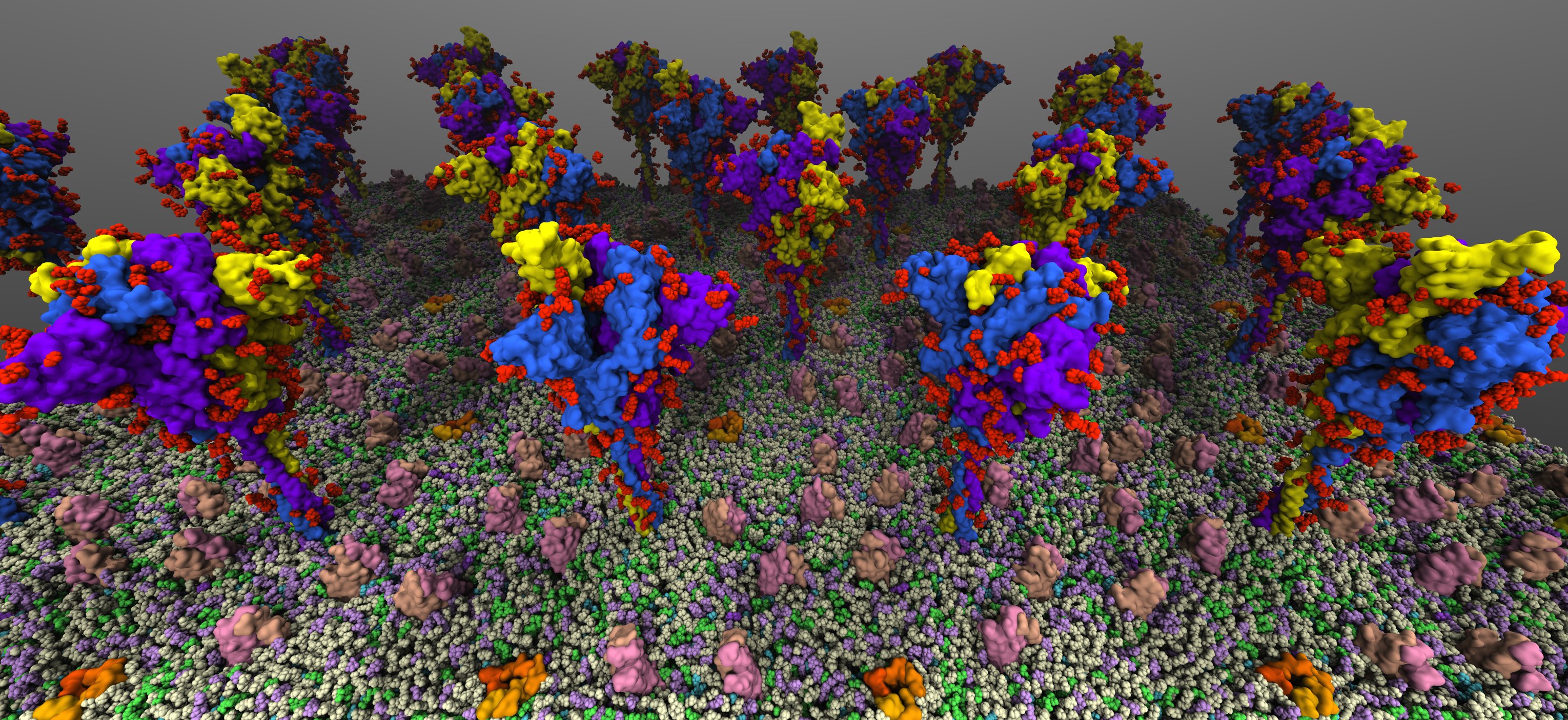
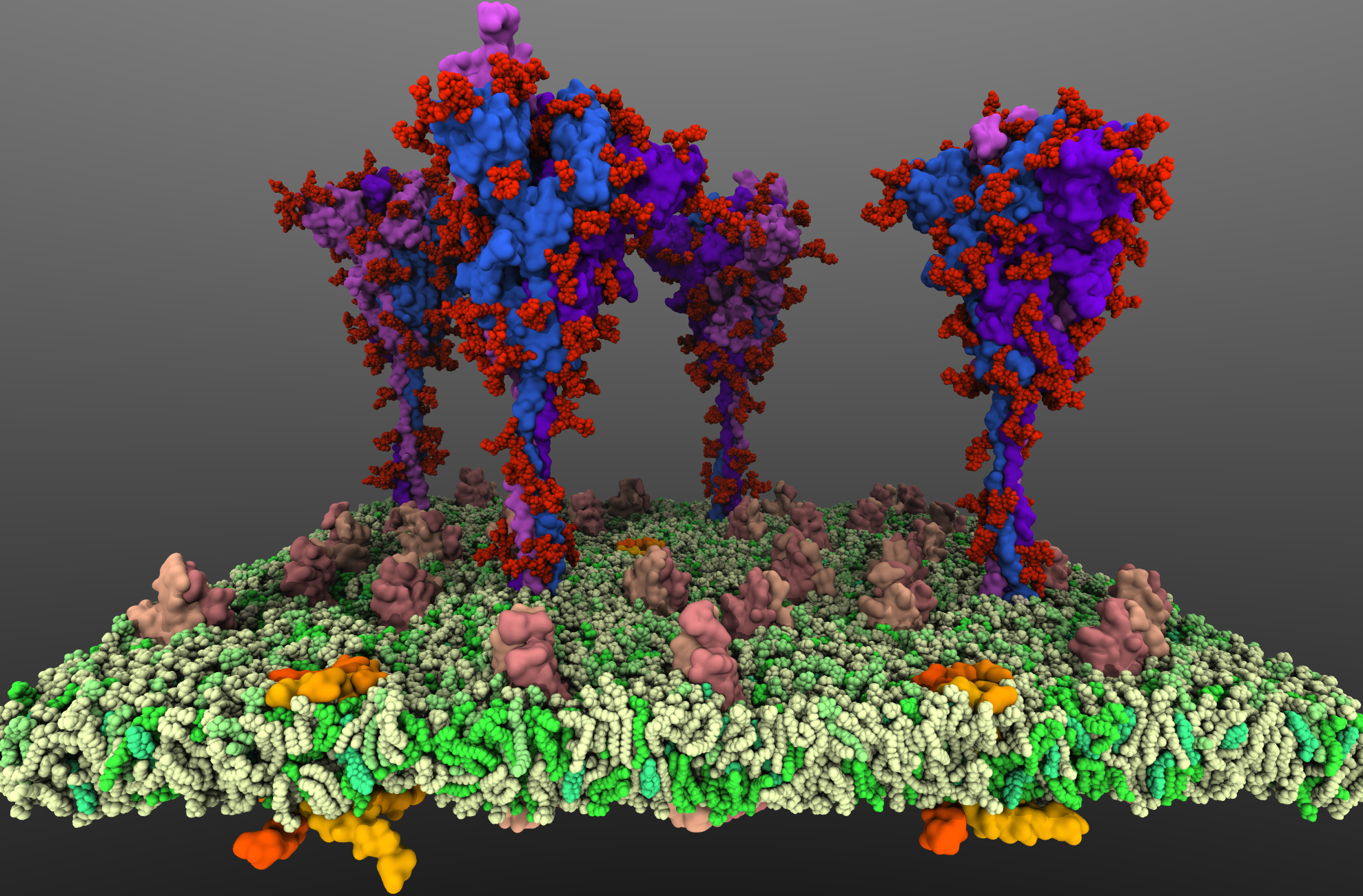
SARS-CoV2 binds to the host cells through its spike glycoprotein (S-protein), making it a key target for therapeutic antibodies and diagnostics. Anchoring of the S-protein in the viral envelope through the transmembrane domain allows it to function as an extended viral antennae for recognition of host cell surface receptors. The construction of the complete membrane-bound S-protein system therefore remains the first and critical step in understanding the working of the most important component of the viral infection
machinery. The recently resolved cryoEM structures of the S-protein lack a number of functionally important regions, including the transmembrane C-terminal domain containing multiple palmitoylation sites and anchoring the S-protein to the viral envelope, as well as critical information about the glycosylation composition of the modified residues suggested to play a protective role against the host immune response. Using a hybrid approach combining homology modeling, protein-protein docking and extensive MD simulations of transmembrane helices, with biochemical data on the palmitoylation sites and the recently reported glycomics data, we have developed a full-length, membrane-bound, palmitoylated, and fully-glycosylated
S-protein model, and tested its stability in short MD simulations. Taking into account the spherical shape of the viral envelope and the high S-protein surface densities observed, here, we aim at characterizing the
conformational dynamics and potential inter-spike interactions of the full S-proteins in their physiological membrane-bound state using extended MD simulations, both in planar membranes replicating
their suggested non-orthogonal lattice configuration, and in a curved membrane patch representative of the native viral envelope shape. The relative protein and glycan plasticity observed from these simulations
will also help us to determine the role of glycosylation in potentially modulating the adaptive immune response to the SARS-CoV2, with major medical implications for the development of effective (multivalent) vaccines against the virus.
This is an active project under the
COVID-19 HPC Consortium.
|
|
Binding Mode of SARS-CoV-2 Fusion Peptide to Human Cellular Membrane
|
Infection of human cells by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) relies on its binding to a specific receptor and subsequent fusion of the viral and host cell membranes. The fusion peptide (FP), a short peptide segment in the spike protein, plays a central role in the initial penetration of the virus into the host cell membrane, followed by the fusion of the two membranes. Here, we use an array of molecular dynamics simulations that take advantage of the highly mobile membrane mimetic model to investigate the interaction of the SARS-CoV2 FP with a lipid bilayer representing mammalian cellular membranes at an atomic level and to characterize the membrane-bound form of the peptide. Six independent systems were generated by changing the initial positioning and orientation of the FP with respect to the membrane, and each system was simulated in five independent replicas, each for 300 ns. In 73% of the simulations, the FP reaches a stable, membrane-bound configuration, in which the peptide deeply penetrated into the membrane. Clustering of the results reveals three major membrane-binding modes (binding modes 1-3), in which binding mode 1 populates over half of the data points. Taking into account the sequence conservation among the viral FPs and the results of mutagenesis studies establishing the role of specific residues in the helical portion of the FP in membrane association, the significant depth of penetration of the whole peptide, and the dense population of the respective cluster, we propose that the most deeply inserted membrane-bound form (binding mode 1) represents more closely the biologically relevant form. Analysis of FP-lipid interactions shows the involvement of specific residues, previously described as the "fusion-active core residues," in membrane binding. Taken together, the results shed light on a key step involved in SARS-CoV2 infection, with potential implications in designing novel inhibitors.
This study has been published in
Biophysical Journal.
|
|
Page created and maintained by Karan Kapoor.