Historical Series: MDFF
Evolution of a Method:
Historical Perspective on Molecular Dynamics Flexible Fitting
By Lisa Pollack
May 2013Revised March 2014
See other related articles by L. Pollack
The game I play is a very interesting one.
It's imagination, in a tight straightjacket. –Richard P. Feynman
“We were extremely naïve,” says Klaus Schulten about a method his group developed and presented to the world in 2007. “And that was our advantage.” The task at hand, which led to this new method, seemed straightforward: take two structures of the same molecular machine, generated from two different kinds of microscopes, and somehow combine them. One type of structure was from X-ray crystallography, and the other cryo-electron microscopy. But, as in real life, there was a catch. The structure at hand was huge, hundreds of thousands of atoms.
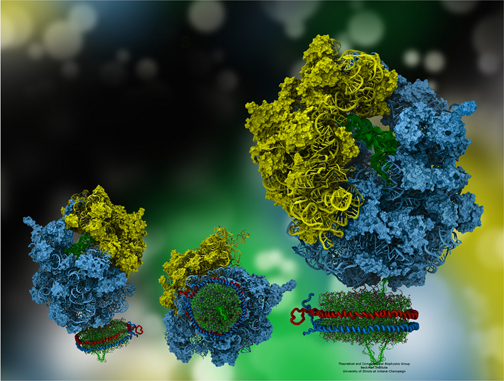
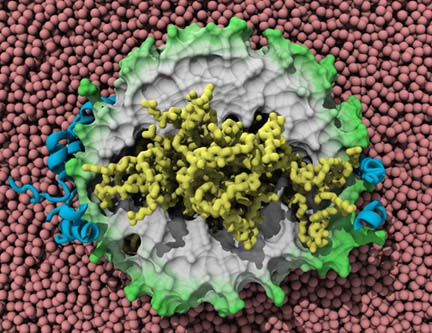
The ribosome, in blue and yellow:
a molecular machine that inspired the creation of the method MDFF.
With the bird analogy, it might seem obvious to take a part of the bird standing still, and manually fit it to the bird in flight: A beak goes here, a claw goes there. While that method was popular when Schulten's group started on their project in 2006, such approaches were not working on the specific quandary they were facing. Not only were the common methods not working, but Team Schulten also had to contend with other constraints: the procedure needed to work on large structures, and due to these huge sizes, had to be fast, computer-wise. The eventual outcome would be called Molecular Dynamics Flexible Fitting, and its genesis required the collaboration of many intrepid minds.
While the creativity of the pioneers of this method was one key to success, their youth was another important element. According to Schulten, it takes an enormous amount of work to build a method from scratch, inevitably involving many paths in the wrong direction, to assiduously construct a solid foundation upon which to build more knowledge in the future. “And young people can do that,” elaborates Schulten, “because it involves so much energy, not only simply intelligence and ability, but also just physical energy.” While young scientists are his capital, Schulten came to this new field rather late in his career; this history examines why Schulten's late arrival to the scene offered him distinct advantages, as well as delineates how young scientists not only laid the groundwork for this method, but also how they continue to elaborate and improve it. While youth plays an important role in establishing and improving this new method, this narrative is filled with a string of coincidences, some of which are very crucial to the successes of the scientists. Although he had been demurring the request for years, a chance encounter at a meeting finally convinced Klaus Schulten to start a collaboration and thus begin his foray into a totally new field. But Schulten's entry into this field came at just the right time, as he caught the beginning of a wave that was slowly building. A random meeting of graduate students in a conference room led to employing a software feature in a completely different way than intended, and would became a hallmark of the new method. And, after glancing at a poster in a hallway in Munich, Klaus Schulten saw for the first time a structure come to life that he had once elucidated, and soon realized that he would revisit it in even greater detail but for totally different purposes.
The whole product of Klaus Schulten's intellectual life has been to comprehend biological organization: “I want to understand the very essence, at least in my mind, of what makes a cell a living cell.” And he wanted to use the tools of physics, mathematics, and chemistry. But simulating a cell is a huge endeavor. In the 1980s Schulten realized he could exploit computers for interpreting large biomolecules; thus began a lifelong odyssey to examine living systems with the computer as a tool to calculate, visualize, and analyze biological systems. And the current method, Molecular Dynamics Flexible Fitting, or MDFF, combines millions of pieces of data from two disparate sources to suggest a structure down to atomic resolution. “When you have something as complex as a macromolecule, made of literally hundreds of thousands of atoms, you need the computer,” explains Schulten about MDFF; “The human mind cannot do this.” But what the human mind can do is use the results of MDFF to illuminate biological processes. And the results are stunning. One of the reasons Klaus Schulten chose to work on this new project in 2006 was so that he could learn more about ribosomes, macromolecules in a cell that are fundamental to life. A ribosome is essentially a “molecular machine” that makes proteins according to blueprints found in DNA. “It stands at the junction between information and action,” summarizes Elizabeth Villa, a researcher in the field. This intriguing machine is so important that the committee in Stockholm awarded three scientists the 2009 Nobel Prize in Chemistry for explicating its structure.
Image of a photosynthetic chromatophore, with light-harvesting proteins in blue and green, some of the many proteins studied in Klaus Schulten's group.
One of the best known facets of this molecular machine is that some antibiotics target bacterial ribosomes to fight infection, leaving human or animal ribosomes unharmed. As malignant tumor growth is, in part, related to a high rate of protein synthesis, ribosomes, or protein factories, may be important in the fight against cancer. In fact, for decades ribosome amounts have been associated with cancer, although the exact mechanism is quite unclear. Recently, researchers have even linked the ribosome to prions, which cause bovine spongiform encephalopathy (mad cow disease) as well as Creutzfeldt-Jakob disease, suggesting that the protein folding activity of the ribosome could be a pharmacological target. While the ribosome may be one key to disease fighting and anti-cancer therapies, the ribosome is beautiful in its own right, as the maker of a cell's proteins. Protein biogenesis or creation is probably one of the most important tasks in living creatures, as proteins perform most of a cell's innumerable tasks. In the human body, commonly known proteins synthesized by ribosomes include: hormones, keratin, collagen, antibodies, insulin, and hemoglobin.
While the above list highlights how critical the ribosome's function is to the human body, there are other fascinating aspects to consider about proteins and their function, which are probably less well known. Wondering how plants convert the sun's rays into a useful energy source via photosynthesis? Look to the plant's complex of light-harvesting proteins. Pondering how a bird could possibly navigate using the earth's magnetic field? Consider studying the protein cryptochrome in its eye. Mulling over how protons move across a membrane in an extremophile? Call on the protein bacteriorhodopsin. Contemplating how to treat Hepatitis C? Perhaps look at its viroporin, a protein the virus produces that acts like an ion channel. Want to understand muscle elasticity? Then titin, a muscle protein, is what to study. Klaus Schulten's research group is actively investigating all of these topics, and each one involves a protein created by ribosomes in living cells.
Because ribosomes and proteins essentially go hand in hand, this history on the method called MDFF explores not only the intricacies of the ribosome, but various aspects that involve proteins as well: Some proteins are designed to pull on molecules, while others simply exist just to stall the ribosome; if a protein's position in a cell is destined to be within a membrane, a protein-conducting channel will guide it there. Furthermore, Schulten's group is using MDFF to look at relevant topics in the world of infectious disease, including HIV and antibiotic resistance, both of which involve proteins.
Schulten is clear to point out that MDFF is a method that suggests a structure to the scientist, as opposed to providing one. But when the structure contains thirteen million atoms, as does the HIV capsid that Schulten and collaborators are determining, the suggestion of a structure is still incredibly valuable. And with that many atoms, the need for a computer becomes paramount, as no other experimental method to date is able to conquer the task. So through a combination of serendipity, youth, curiosity, and perseverance, a new technique arose, which is now starting to attract the attention of structural biologists, too. In summary, the name “Molecular Dynamics Flexible Fitting” says it all, for as Schulten himself contends, “Flexibility is what we brought new to the game.”
Joachim Frank Calls it “The Queen of all Microscopes”
Biology today would be unthinkable without the invention of the electron microscope.
These words were first written by Joachim Frank in a 1996 book, and it is with him that we begin the account of the history of MDFF, a technique whose emergence was based on some electron microscopy data on the ribosome that Frank had generated. When Frank asked Schulten to collaborate, Schulten realized he knew little about the ribosome, and even less about combining X-ray crystallography with electron microscopy maps, and hence he would be taking a big risk. But Schulten was intrigued, and certainly open to risk. He had immense respect for Frank, who, like Schulten, was trained as a physicist but studied biological systems. Schulten also realized the ribosome was an awesome cellular machine that merited his attention. As the focus of Schulten's group was on simulating large biological systems using techniques and software developed in-house, he knew also that he had, in principle, the methodology and know-how to conquer a system as big as the ribosome. “We had a really extremely important biological problem, and we had an absolutely outstanding investigator,” recalls Schulten about his decision. “So I think I would have been a scientific low life if I said I wouldn't consider it.” But their road to collaboration was not straightforward.

Joachim Frank, who images ribosomes with the electron microscope. Photo used with permission from Joachim Frank.
Joachim Frank did not start out with the explicit goal of becoming an expert on the ribosome; this enigma he would pursue in what he refers to as the second stage of his career. Completing his graduate studies in Munich in 1970, Frank started thinking about how to make use of the electron microscope in more inventive ways than it had ever been used before. As far as microscopes go, many are familiar with the light microscope, which uses rays of visible light to probe a structure and magnify it. As its name would suggest, the electron microscope uses beams of electrons instead of visible light; its advantage is that it can magnify smaller structures than a light microscope, and reveal objects as tiny as atoms. Frank, in his aforementioned book, Three-Dimensional Electron Microscopy of Macromolecular Assemblies (1996), makes an elegant case for why the electron microscope (appearing on the scene during the 1930s) so revolutionized biology.
The detailed architecture of the cell and its numerous organelles, the structure of viruses, and the fine structure of muscle are all beyond the reach of light microscopy; they came into view for the first time, in spectacular pictures, as this new instrument was being perfected (1).In the first part of his career, Frank worked diligently to perfect a technique known as single-particle reconstruction, and he is considered one of its pioneers. Frank revealed that early on he wanted to explore imaginative ways to use the electron microscope because he realized it could capture molecules in a functional state, that is, as they performed their work. This is in contrast to the structures provided by another microscopy called X-ray crystallography. To capture a structure via X-rays, the object in question must be made into a crystal, that is, identical copies of it must be arranged in periodic fashion and tightly packed together; only after the object is crystallized can X-ray diffraction be performed and the results tediously translated into a structure. This form of microscopy does have the advantage of yielding structures at atomic resolution, thus providing a bit more information than electron microscopy. But this leads to certain consequences: “So the dichotomy is between having molecules in a crystal formation or aggregated in some kind of a helical fashion, and on the other side having the molecules completely free standing,” states Frank about the two microscopies, “because only in the latter case will they be as they are in the cell.”
What Frank was able to achieve, harnessing the electron microscope in a new fashion, took him nearly two decades. Below is just a summary of how he perfected “Single-Particle Reconstruction,” but a more detailed account of his journey can be found in a thorough historical article Frank wrote in 2009.
Around graduate school in the late 1960s he started thinking about how to turn two-dimensional scans from the transmission electron microscope into a three-dimensional image. Regarding the molecule in question, with many thousands of copies laying on a grid, Frank sifted through a multitude of 2D images of each randomly aligned “single particle.” The hard part was using the myriad 2D images to “reconstruct” a 3D density map. And for this he developed a computer program, called SPIDER, which debuted in 1978 and is still in use today. (The acronym stands for System for Processing Image Data from Electron Microscopy and Related Fields.) Around this time Frank realized that the ribosome was an excellent candidate for testing the methods he was continuously refining. When combining 2D images from multiple ribosomes, the stability of the ribosome was key since Frank was processing so many copies of the structure to create his 3D map. The size and contrast of the ribosome also made it attractive.

The electron microscope used in Joachim Frank's lab at Columbia University. Photo used with permission from Joachim Frank.
When a technique became available that allowed samples under study, like the ribosome, to be captured in their original form (for example, staining of samples was not necessary), Frank adopted it in his lab. The technique–cryogenic electron microscopy–meant that samples were frozen before being put under the microscope, but this instantaneous freezing captured the ribosome in a functionally meaningful way. The ribosome imaged this way was very different from what X-ray crystallographers were able to achieve, for in a crystal the system is confined to only certain geometries. However, Klaus Schulten is clear to point out that sometimes the crystallized particle functions normally. “Now some systems–which are rare, but they absolutely exist–when they are in the crystal they are as good as when they are outside of a crystal,” Schulten reiterates. “They do everything in the crystal the way they do it outside. It's rare, but it happens, absolutely.”
By this time Frank was intrigued with the ribosome as more than just a poster child for the electron microscope. “It is such a complex molecule, and it has such a central role in the cell and in life,” responds Frank when asked why this molecular machine fascinates him. He is also quick to point out that many of the resources of the cell go into making ribosomes, so much so that a cell is perpetually making ribosomes. In fact, in a growing yeast cell, about 2,000 ribosomes are made every minute. “The ribosome has such a central importance and such a central role,” Frank says, “that putting the majority of all the energy into making it makes a lot of sense.”
By the early 1990s Frank was now implementing cryo-electron microscopy on the ribosome and slowly getting better and better resolution as the decade progressed. At the same time other scientists were working on trying to get the structure of the ribosome via X-ray crystallography, but having little luck actually forcing the ribosome to form compact crystals that provided adequate resolution. In retrospect, Frank recalls the hope that “around 1995 I will have the idea that we might even win the race in getting the structure with electron microscopy rather than with X-rays.”
But by the late 1990s scientists were finally able to make suitable crystals of the ribosome, and Frank relates that the year 2000 marked the fast succession of many published X-ray crystallography structures. He also notes that the whole idea of what electron microscopy could be used for was switching over. In cryo-electron microscopy, the ribosomes are in the course of doing their biological function right before they are frozen. One way to look at the ribosome is as a molecular machine that goes through several states to perform its work. Each functional state may involve slightly different ribosome geometries. As the geometry of the ribosome is critical to understanding each state, a method that can capture all the different states could better elucidate function–and cryo-electron microscopy is exactly the method that could yield images of the ribosome in its intermediate states.
In 2000 Frank published a paper in which he found a ratchet-like motion happening to the two subunits of the ribosome as they assembled a protein, capturing “a molecular machine in motion,” wrote Frank in his 2009 history article. Soon after the ratchet motion paper, X-ray structures of the ribosome appeared on the scene. With X-ray crystallography now yielding structures down to atomic resolution, Frank realized he could use all this crystallography data to interpret electron microscopy maps he was generating. Specifically, he wanted to see the molecular reorganization that occurred within the ribosome as it went from one state to the next in the course of its function.
Sometime in the early part of the new millennium Frank was at a meeting, and a colleague, Harry Noller, told Frank about a method to fit X-ray crystallography data to cryo-electron microscopy maps. Called real-space refinement, the approach was being honed by Mike Chapman at Florida State University. Frank was able to collaborate with Chapman, even though the structure Frank gave Chapman was larger than anything Chapman had ever worked on before. “Real-space refinement is a way of essentially breaking the structure into pieces, more and more pieces,” explains Frank. “And the breaking points are always put where there are natural points of flexure.” While Frank and Chapman and colleagues did get the method to work, and published in 2003, this specific foray into fitting X-ray crystallography data into electron microscopy maps left Frank aware of the limitations of the method called real-space refinement.

The computational microscope, a tool that yields information available with no other form of microscopy.
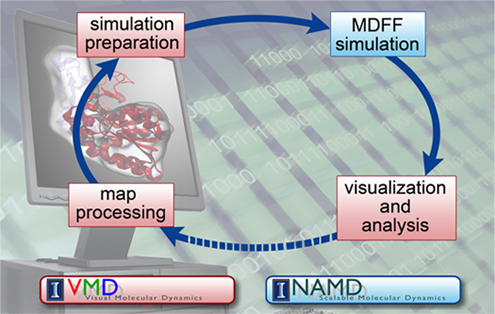
Around this time, while Frank was working on real-space refinement, he saw Klaus Schulten at a meeting, and Frank realized that now was his chance. Over the years Frank had come to know about Schulten's work, and the prominent role Schulten had in the world of molecular dynamics. Molecular dynamics is a simulation technique that supplies a scientist with a look at the motions, or dynamics, of a molecule under study, and furnishes insights into its behaviors that are impossible to capture with any other form of microscope. Schulten's group had refined molecular dynamics over the years to work for larger and larger systems, and for longer and longer time scales. Not only did Schulten's group specialize in a program to run molecular dynamics in parallel, NAMD, but his group also worked on a software program called VMD, which is both a visualization and analysis tool. The pair of companionate tools, NAMD and VMD used together, frame what Schulten calls the computational microscope. Using the information from math, chemistry, and physics, combined with software and hardware, this non-traditional microscope imparts users with a feel for a molecule as a dynamic structure in motion.
The work Schulten did, especially on simulating large systems, appealed to Frank, who also studied a large system as it performed its key function, namely the ribosome and translation. Protein synthesis is extremely complicated and involves many steps, and one of them is translation. In order for the ribosome to make a protein, it needs a blueprint for the order of the individual parts (amino acids) of the protein it assembles. This blueprint resides in a cell's DNA, but the actual blueprint the ribosome can read is brought to the ribosome as a strand called messenger RNA or mRNA, which is related to the instructions found in the DNA. Simply put, the process by which the strand of mRNA attached to the ribosome is used to assemble a protein piece by piece is called translation. “I figured that in order to understand protein synthesis one would have to essentially be able to simulate some of the key sub-processes, or even simulate translation in its entirety,” recounts Frank. But in all of Frank's attempts to collaborate with Schulten, Schulten had not followed up–until Frank saw him face to face, circa 2003/2004. Schulten eventually assigned some students to the problem of how to fit X-ray crystallography data to cryo-electron microscopy maps of the ribosome. And so began “The Ribosome Project” in Schulten's group, and his students would be working mainly with Joachim Frank and his graduate student Kakoli Mitra on brainstorming ideas for fitting.
Schulten assigned two young scientists to the ribosome project, and asked a third as well; but this third person demurred, even though she was fascinated by the ribosome and even though she already had a connection of sorts with Joachim Frank. Instead of refocusing all her efforts on the ribosome project, graduate student Elizabeth Villa told Schulten she would just take on a role as a consultant, as she was almost at the end of her PhD. But eventually the project on the ribosome would become a central focus of Villa's attention as well.
Ribosome Rocks
When Elizabeth Villa was a new graduate student at the University of Illinois, she did a rotation in Klaus Schulten's lab roughly in 2001, and told him about how the crystal structures of the ribosome had just been published, and how it might be a project he would want to consider. According to Villa, Schulten always wanted to work on the ribosome, and saw it as an excellent challenge for his methodology.

Elizabeth Villa, who worked in the Schulten group 2001-2008.
During this time, Villa was taking a course, “Statistical Physics applied to Biological Information and Complexity.” In one of the essays required for the course, Villa wrote a piece entitled “Ribosome Rocks,” a play on words referring to the motion of the ribosome as it ratchets. Cleary Villa had studied Joachim Frank's 2000 paper where he describes the ratchet motion observed in electron microscopy, as she makes reference to his publication in her essay.
Meanwhile, Joachim Frank's publication about the ratchet-like motion of the ribosome debuted in Nature in 2000. “Months later, I searched the internet, because that was sort of a new pastime, surfing the internet,” reminisces Frank, “to see whether there was anybody who made reference to it or somehow used the information.” He came upon Villa's essay “Ribosome Rocks,” and hunted down her email address and sent her a message of encouragement. Villa was thrilled, to say the least. She was so excited that she ran to Schulten's office to report the good news. Schulten suggested Villa include in her response a note that Schulten sent his regards and that Schulten was going to look into working together.
Assembling a Team for the Ribosome
Although Schulten had been considering collaborating with Frank as early as 2001, as the above anecdote suggests, it took about four or five more years before anything concrete happened. So when Schulten finally decided the time was right to work with Frank, it seemed obvious to ask Elizabeth Villa to join the project. He assigned two others as well, new graduate student Leonardo Trabuco and postdoc Emma Falck, while Villa still insisted she would be only a consultant. Joachim Frank assigned graduate student Kakoli Mitra on his side.
The first task for Trabuco and Falck was to learn a current method for fitting X-ray crystallography data to an electron microscopy map, a new type of fitting known broadly as flexible fitting. For this, they went to a workshop in Houston given by Willy Wriggers, actually a former member of Schulten's group, who had written software for fitting methods. Trabuco and Falck then decided to use Wriggers's method on their ribosome project. But a wrinkle developed, based on the components of the ribosome. This molecular machine is made up of not just proteins, but a type of molecule called ribosomal RNA or rRNA; for example, in bacteria, the ribosome consists of roughly 65% rRNA and 35% proteins. A fitting technique that worked on the ribosome would have to succeed not just for proteins, but also for the other main component of ribosomes, namely RNA. Trabuco and Falck and Villa discovered, upon working with Wriggers's methods and software initially, that the flexible fitting scheme they were using failed to fit the RNA correctly. They eventually realized it was because while the methods were optimized to fit proteins, they did not take into account RNA. The team was at a standstill. They realized they would need to create something from scratch, yet no one suspected that the answer was right under their own roof.
An Unexpected Solution
Villa, Falck, Trabuco, and Schulten started brainstorming for other ways to fit the X-ray crystallography data into a cryo-electron microscopy map. Most of the ribosome team was at a dinner one night, and another person attending was also in Schulten's group, graduate student Jordi Cohen. He was obviously thinking about the dilemmas the group faced in the ribosome project, for not long after that, when Trabuco and Villa and Cohen were in a conference room together, Cohen suggested they might want to use a new feature in NAMD, called Gridforces. “And I remember going, 'Yes! That's it! It's that simple,'” exclaims Villa, “because all the things we were thinking were so much more complicated and convoluted.”
Gridforces appeared in the Schulten group from a project totally unrelated to MDFF. Now a professor of physics at the University of Illinois, Aleksei Aksimentiev was a postdoc in Schulten's group and in 2004 wrote an algorithm to move along his work studying membrane transport. Aksimentiev and Schulten had been investigating how the confinement of the alpha-hemolysin channel affects molecules of DNA being driven electrically through the channel. Initially, they employed a specific mathematical description for exerting confining forces on DNA, but around 2006 a graduate student of Aksimentiev's, David Wells, developed in NAMD a general algorithm for confining forces, which they termed Gridforces.
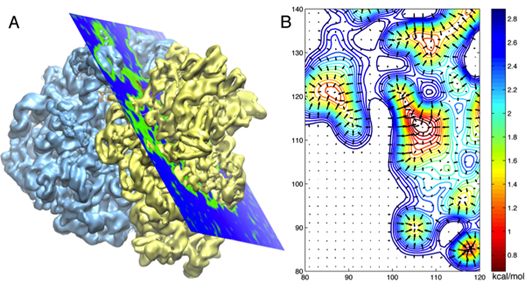
An electron microscope map of the ribosome on the left, and a 2D cross section of the potentials derived from the map in A.
The Schulten group then decided to employ Gridforces to drive the ribosome into the cryo-electron microscopy map. They were literally pushing the structures determined at atomic resolution into the map in their simulations with Gridforces, in particular, moving atoms to areas that are more dense, and away from low-density areas. (For hardcore molecular dynamics users, what the procedure in MDFF does is to define an extra potential to augment the usual molecular dynamics potential.)
Schulten's various conceptual descriptions of what goes on in the fitting are very helpful, because it highlights both the ideas of flexibility and naiveté. Consider a soccer match, and an outsider who wants to figure out the rules of the game, but subject to the following conditions: The details of all the players are clearly captured when they are standing still before the game starts, singing their anthem and being photographed, and one can distinguish clearly the eyes, hands, and feet of each player. But then play starts. What electron microscopy is doing is freezing the players in a state while they are moving on the field. But it is much more difficult to get as crisp a picture of all the players in motion and moving so fast than it is to get a picture of the players standing still before the game. Now to figure out the rules of soccer, the curious outsider has to match up the players in the still photo to the less-resolved image of the players in motion.
The kind of fitting that was around when Schulten's group took on the ribosome project was basically taking sections of the crystal structure and fitting it piece by piece into the electron microscopy data, that is, locally fitting things around. But this way was difficult and did not always work. Elizabeth Villa even refers to it as butchering. In the soccer analogy, it would be taking each part of a still player's body, the feet, the hands, the arms–placing body parts instead of whole bodies. But the group realized that the ribosome had so many parts that this piecemeal approach was impossible; plus the ribosome was too large and it would take too long.
The eventual approach, MDFF, also needed to keep in mind the overall chemical structure of the molecules it was fitting. For example, one would not take an arm from a still player and put it in the middle of the field. The curious observer needs a sense of how the human body functions, i.e. that an arm always has to be attached to a shoulder. In the same way MDFF knows about the chemical structures and mechanical properties, e.g., the flexibility, of macromolecules.
But in Schulten's mind, the innovation that framed MDFF was the group's naiveté; “Our naiveness was: let's just assume any part of the atoms can go anywhere, because our molecular dynamics program is so good that when we let the protein go, the molecular dynamics program will keep everything intact enough that we don't screw up the macromolecules with their chemical structure and mechanical properties,” Schulten clarifies. Basically the molecular dynamics allowed the ribosome to deform itself from the observed crystallographic structure, which does not correspond to a functional state, into the electron microscopy map that does represent a functional state. And it does so by permitting only conformational changes that can be reconciled with known chemical structures and mechanical flexibilities.
Refining the Method
With Gridforces now a tool in the arsenal to try, the team still had tons of work to do. By this time Villa had evolved to more than just a consultant, swept in by the excitement of the ribosome, and Emma Falck had left the project. The duo, Villa and Trabuco, actually had a deadline that was looming large. In truth, the entire group of Schulten's was in high-stress mode. The renewal for Schulten's research center, now called the Center for Macromolecular Modeling and Bioinformatics, funded by the National Institutes of Health, was on everybody's mind in 2006. Renewal involved writing an extensive document highlighting all the accomplishments in the last five years, proposing projects for the following five years, and a site visit by a committee in March of 2007. Villa explains that while all this was going on, Schulten also wanted team members to complete at least twenty percent of the research on all things proposed as future work, so Schulten could present this peek into ensuing work for the visitors.
With Gridforces, Villa and Trabuco set about testing it for fitting the proteins in the ribosome. This involved a lot of work, writing their own scripts in VMD, and making heavy use of NAMD as well. “For proteins it worked, in a picosecond! It was incredible,” Villa declares. “I never actually expected it would work that well.” But their celebration was short-lived. The next big test was the RNA in the ribosome. The previous methods Trabuco had used for fitting RNA into the ribosome had failed, so the pair knew this obstacle was sizable.
With the approach the pair took, now using Gridforces for fitting, they were essentially applying outside or artificial forces in the procedure. One consequence was that these artificial forces pushing the structure to the map could distort the structure itself and blow up the simulation. Schulten and his students had to invent some cautionary measures to basically make sure that key structural elements of the ribosome stayed intact and did not distort during the fitting. These “cautionary measures,” or attempts to preserve structural elements, became known as restraints, and the incorporation of restraints, not just for RNA, into the MDFF methodology played a big role in its future success.
Help from Within
The Theoretical and Computational Biophysics Group is a large one, and filled with diverse projects in biophysics, parallel programming, and molecular visualization. The skillsets of the scientists in the group range from math, physics, and chemistry to software development. Many, in fact, need to possess all those skills at once. And each scientist's project directory is readable by the whole group. This kind of environment, where ideas are shared and the collective knowledge and skills of the group members are vast, is one of the reasons Trabuco, Villa, and Schulten created MDFF so successfully and relatively quickly. “People are encouraged to go look at examples of what others do, which is great, as you can learn very quickly,” Villa explains about the group's atmosphere. “So you can always build on stuff that other people do.” The fact that two software products, as well as the very developers of VMD and NAMD, were at their disposal makes it clear that the broadly available resources in the Schulten group were a sine qua non for MDFF's success.
Leo Trabuco did an inordinate amount of coding and script writing to get MDFF to actually function. He remembers how he struggled with the first version of the protein restraints. “I was doing a lot of hacks to change the force field, and it was getting very convoluted,” Trabuco recalls. “A lot of pre-processing and redefining the force fields so that we could set up restraints for conformational changes in the protein.” Down the hall worked Jim Phillips, the main developer of NAMD. Trabuco recalls talking to Phillips about his pursuits with the fitting and the protein restraints, and how excited Phillips seemed with this new procedure, MDFF. Trabuco also talked about wrestling with the protein restraints. Unbeknownst to Trabuco, Phillips went back to his office and, in barely a day, tweaked some code in NAMD. “Jim came back and said: I wrote a feature so that you can just give an input file and you can have all these restraints in NAMD. You don't have to do all of these other things,” notes Trabuco, who was not familiar with NAMD code and appreciated the easy, and generous, solution by Phillips.
While Jordi Cohen was instrumental to MDFF's success, suggesting the use of Gridforces, Trabuco also took help from some code Cohen had written as well. Cohen, in his own work, needed to create three-dimensional maps of molecules, and had written code both in VMD and stand-alone C++ programs to further his mapping project. Trabuco extended this feature in VMD, but that's not all. For MDFF Trabuco had to do a lot of map processing when working with the electron microscopy data. He first used external programs for this. “And then slowly I started using Jordi's code and extended it to implement all the functions that we needed,” says Trabuco. “For example, filtering of the map, clamping negative values, calculating 3D correlation maps, and a lot of things like that.”
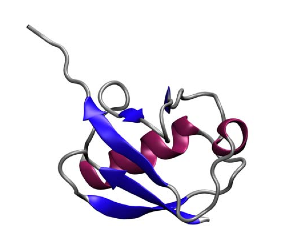
Protein secondary structures: beta sheets in blue, alpha helices in red. Ubiquitin, another protein Klaus Schulten's group studies.
Trabuco also received help with preserving protein secondary structures from another group member. Proteins are chains of amino acids. Amino acids have a similar underlying structure and are distinguished from each other by their side chains. The primary structure of proteins is related to the sequence of amino acids. But proteins have what are called secondary structures, and this is related to common structural elements that appear when a protein is folded. Alpha helices and beta sheets are secondary structural elements, stabilized by hydrogen bonds, and a single protein can have many sheets and strands identifiable with a visualization tool like VMD. Beta sheets are somewhat akin to folded sheets of paper, and Trabuco realized that when fitting his proteins, nothing was holding the sheets together, as he only had restraints for dihedral angles in place. He then implemented hydrogen bond restraints to fix this problem, and based this on code that another group member had written, namely Peter Freddolino.
As delineated above, the shared resources in the Theoretical and Computational Biophysics Group were important for Villa and Trabuco as they struggled to develop a fitting method. But Klaus Schulten, the group's founder, points to another aspect of his group that is also important: its long-standing collaborations with experimentalists. Schulten's first job in 1973, straight out of graduate school, was at the Max Planck Institute for Biophysical Chemistry, where he was basically the only theorist in a group of dozens of experimentalists. That experience deeply shaped him, and not only did he learn an immense amount about experiments, he also learned to speak their language as well. As a result, Schulten's group has collaborated with experimentalists innumerable times since 1973. “We learned to understand them, and they learned to trust and respect us,” Schulten clarifies. “So I think our ability to work with experimentalists was something that very few groups have to the extent that we have it.”
Elizabeth Villa became the main communicator with Joachim Frank's lab. “I really liked the technique [electron microscopy, or EM], and I already sort of had a relationship with Joachim, and so I dove into all these physics books, to really try to figure out what these data were,” Villa reflects. “This was very useful for me, because when I started doing EM myself, it was nothing like in the books–it was much harder.” Villa would actually work with the electron microscope after departing Schulten's group.
One of the main sources for Villa was Joachim Frank's 2006 book, Three-Dimensional Electron Microscopy of Macromolecular Assemblies (the first edition published in 1996). When Villa and Trabuco would get stuck in their undertakings, Villa would often get in touch with the people in Frank's lab to help her. She would go through graduate students, postdocs, the microscopist, and the processing person, often without answers. “It was always back to Joachim and he would say: Check page 213, second paragraph, that's the equation that you need,” Villa recounts. “So I think it was a lot of fun for him too, to sort of recap all this knowledge that he had, and apply it to something new and try to figure out things.”
Ask for the Shiniest Map
At the end of 2006, Villa and Trabuco, after much exertion, decided that they had a respectable working model of MDFF, and Schulten decided to present it at the upcoming critical site visit, which would play a huge part in determining if Schulten got a five-year renewal for his center. Trabuco and Villa made great strides on MDFF between proposal submission and site visit, a period from October 2006 to March 2007, and this progress was a pleasant surprise for Schulten. The MDFF method would become a focal point of the site visit. (Schulten, by the way, did secure the NIH renewal.)
Villa and Trabuco had been using sample maps from Joachim Frank when they were first trying to get the method to work. When Schulten was given a preview of MDFF and realized the pair were on to something, he told them to request a real map with a scientifically relevant problem. “So this was Klaus's suggestion,” Villa recalls. “He was probably very smart in saying: 'Why don't you ask Joachim Frank to give you his shiniest map?'”
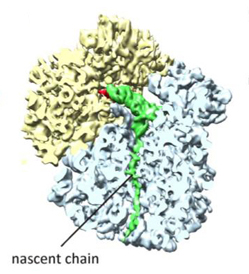
The ribosome, in blue and yellow, and in red, the elongation factor, which acts like a gate.
Villa contacted Frank, and relayed to him some sample movies of the fitting procedure, and Frank was excited by the progress. When she then asked for his best map, she got it, but with a caveat. While Frank said he was happy to send it to Schulten, he said he had given the map to two other people who were also going to try and generate atomic models of Frank's electron microscopy map with their own fitting procedures. As Villa tells it, “We thought: these are people who know what they're doing and evidently they've been thinking about it for awhile.” Villa realized it was a case of “may the best scientist win.”
Frank apparently liked what Schulten's group had done, for when they presented the fitted structures to Frank, he was very excited and told them they needed to write up the results for publication. The map he gave them contained not only the ribosome but other structures and proteins attached to it as well, which were important for elucidating the elongation cycle. Very simply put, as a ribosome makes a protein, this chain of peptides grows amino acid by amino acid, and this step-by-step lengthening of the nascent chain is called the elongation cycle. A critical protein in this process is the elongation factor Tu (i.e. EF-Tu) that accompanies a tRNA along with a particular amino acid. Using Molecular Dynamics Flexible Fitting, the Schulten-Frank team was able to obtain an atomic model of the system during key steps in the elongation cycle and better understand the behavior of the elongation factor as well. The elongation factor, EF-Tu, is relatively far away from where the genetic information in the form of mRNA is being read in the ribosome. Results revealed that upon recognition of the fundamental genetic segments, the codons, by the tRNA, a gate is opened in the EF-Tu; this gate allows water molecules to access a substrate attached to EF-Tu and a signaling reaction then occurs, leading to elongation of the nascent protein through addition of the correct amino acid. The results were published in the Proceedings of the National Academy of Sciences in 2009. Thus would begin the fruitful collaboration between Klaus Schulten and Joachim Frank that has produced many other publications.
Stalling for Progress
While Schulten began to seek other systems to apply the burgeoning MDFF to, he would never dream that a system he first worked on in 1997, and which lived vividly in his imagination only, would spring to life upon contact with a new collaborator on an overseas trip.
The first collaboration between Klaus Schulten and Roland Beckmann involved the protein TnaC (in green) as it stalled the ribosome that was assembling it.
The Gordon Research Conferences are run by a non-profit organization that aims to provide forums for cutting-edge research in the biological, chemical, and physical sciences. In June 2008 one of the conferences that year was “Technical Advances and Recent Breakthroughs in Three-Dimensional Electron Microscopy,” and Leonardo Trabuco went to Italy to attend, to learn as much as he could and also seek collaborators. One of the speakers in the session was an electron microscopist from Munich, Roland Beckmann, who actually trained in Joachim Frank's laboratory. That day Beckmann spoke about his work with newly forming proteins (nascent proteins) and their interactions with the tunnel in the ribosome through which they exited.
Trabuco was intrigued. “We got together,” Trabuco says about meeting with Roland Beckmann at the conference. “And he presented his work to me and explained everything. And I was blown away by the resolutions.”
Trabuco realized that the maps of the ribosomes that Beckmann produced were the highest resolution Trabuco had ever seen. While some scientists got better resolutions from their electron microscopes, those were for structures of high-symmetry, like highly-symmetric viruses, and in those cases MDFF was not necessary there, according to Trabuco, because scientists could use ab initio or crystallography-based models to build atomic pictures of their systems. Not only was the resolution excellent, but Trabuco also realized that the biology of the nascent protein in the ribosome exit tunnel was exciting.
“So then I went back to Urbana and I went to Klaus and said: 'You have to get this guy. You have to convince him to collaborate with us. I think that this is the best thing that can happen to MDFF.'” Schulten needed little persuasion and so a collaboration was born.
In the first Schulten-Beckmann collaboration, which led to a 2009 paper in Science, the mechanism of stalling was elucidated in the bacterium E. coli. As discussed before, the ribosome makes proteins; as the proteins are being formed, they exit through a tunnel in the ribosome. Certain nascent proteins are “Born to Control” as Klaus Schulten has coined it, and they actually can stall the ribosome that is assembling them. But to what end?
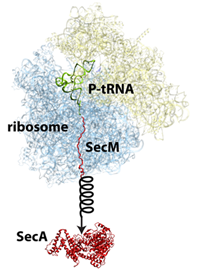
The protein SecM in the ribosome stalls its own production so that another protein called SecA can be made.
But this was not the only project on stalling that Schulten's group worked on. Another protein in E. coli, called SecM, has an intrinsic stalling capability, whereas TnaC stalling described above is dependent upon tryptophan levels. The job of SecM is seemingly only to stall the ribosome making it, so that another protein encoded further down the messenger RNA can be produced by a second ribosome. In this fascinating process, stalling by SecM leads to the production of a protein, SecA, whose job is to pull proteins across membranes. When enough of this puller protein is made, the puller protein itself actually tugs on the SecM attached to the ribosome to cease stalling. While MDFF was employed to develop an atomistic model of a SecM-stalled ribosome, extensive molecular dynamics simulations were run as well to clarify this mechanism. While doing MDFF to get an atomic-resolution structure is one thing, this work, done by Schulten and his group members James Gumbart and Eduard Schreiner, also highlights that further scientific studies can be done on the structures obtained with MDFF. The SecM study, published in 2012, highlighted how SecM can interact with a segment deep in the center of the ribosome to halt its own stalling. Furthermore, Schulten's group simulated the pull on SecM to reveal how SecM might become unstuck.
Believing is Seeing
While vacations are usually associated with some form of relaxation, for Klaus Schulten, during one particular trip his heart skipped a beat. Once or twice a year since 1988, when he began living in the United States, Klaus Schulten has gone back to Munich. Both he and his wife have many collaborators in that area, and the couple likes to combine work and relaxation, especially in the summers when they can hike in Bavaria. To know Klaus Schulten is to realize that he only ever takes “working vacations.”
Klaus Schulten saw for the first time a nanodisc (bottom) on the wall of Roland Beckmann's laboratory, in an electron microscopy map very similar to this one.
The nanodisc is a bioengineering device developed at the University of Illinois at Urbana-Champaign by biochemist Stephen Sligar, director of the School of Molecular and Cellular Biology. But its early roots go back to another Illinois researcher named Ana Jonas and her work on lipoproteins in the 1980s and 1990s. Lipoproteins are found in the blood of vertebrates and they are responsible for transporting fatty acids, lipids, and cholesterol. The two types of lipoproteins most famous are probably HDL and LDL (which stand for high-density lipoprotein and low-density lipoprotein, respectively). HDL is often referred to as “good cholesterol” in the doctor's office.
Ana Jonas, a biochemist, was specifically interested in HDL, and in fabricating them in her lab for analysis. In general a lipoprotein is a globular structure that has a shell made of lipids, an inner core of fats, and proteins on the surface of the molecule called apolipoproteins. These apolipoproteins on the surface are essentially a marker for the liver so that it can recognize and take in HDL, but they also function to recognize and collect the lipids and cholesterol that will be stored in the center of HDL. Shown is an image of an HDL and its components, an example of one kind that has collected cholesterol in its center.
One image of HDL, high-density lipoprotein, with its apoliproteins in blue and cholesterol ester in yellow.
In the mid 1990s Ana Jonas approached Schulten, asking him to help her visualize the HDL with its surface apolipoproteins. Schulten worked on the project, generating a model of the HDL, but he never liked the final product they sent to the publisher. And it turns out, with good reason. Jonas had a collaborator who gave her very specific information about the configuration of the apolipoprotein surrounding the HDL. But it turned out to be incorrect. And eventually Schulten and the rest of the scientific community realized the mistake.
When Steve Sligar dreamed up the idea of a nanodisc, people thought he was nuts. But he wanted to engineer a nano-system to hold a membrane protein, to basically fool a membrane protein into thinking it was in its natural environment. When a ribosome makes a protein, the protein may be destined for living inside the cell in its aqueous environment, it may be peripherally attached to the cell membrane, or it actually may go to live inside a cell membrane. Proteins that reside inside a membrane, called integral membrane proteins or integral proteins, are very difficult to study because they become inactive and essentially die when outside their native membrane. For illuminating the first structure of a membrane protein, three scientists were given the 1988 Nobel Prize in Chemistry for work performed in the 1980s. Hence detailed structures of integral membrane proteins have not been around for a long time, only beginning to appear in the late 1980s. Strategies to further the study of membrane proteins are critical, for as Schulten likes to point out, about a third of all the proteins made in a cell are membrane proteins.
Cell membranes are made up of lipid bilayers. Lipids have heads and tails, and in the bilayer the tails of the top layer and bottom layer are pointing inward and essentially meeting each other. Sligar realized he could create a lipid bilayer and wrap around it scaffold proteins to stabilize it and hence create what he came to call a nanodisc. He based this on the work Ana Jonas had done, fabricating HDL particles in a lab. A very simple schematic of a nanodisc is shown to highlight the lipid bilayer surrounded by two proteins. With the golden hand of Sligar, these devices essentially self-assemble in the laboratory and they are so small, with a typical diameter of about ten nanometers, that they are considered nanoscale devices. Sligar worked for years perfecting this device, cutting away the scaffold protein to its absolute minimum, taking off the receptor part that docks to the liver, until he had his recipe for success around the advent of the new millennium.
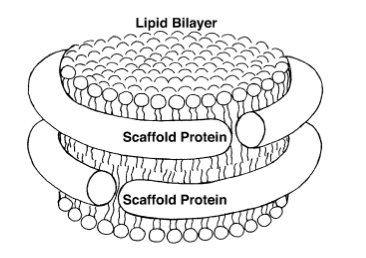
A basic image of a nanodisc, whose bilayer consists of lipid tails pointing inward.
Schulten and Sligar, longtime faculty friends at the University of Illinois, then started a collaboration around 2003, Schulten tasked to simulate the nanodiscs that Sligar created in his lab. Schulten had a graduate student, Amy Shih, who did molecular dynamics simulations, looking at various sizes of the scaffold proteins that encircle the nanodiscs. The computer simulations revealed that the self-assembling particles formed disks, so this was one step in proving the shape of the nanodiscs. To further this notion that the structure was disk-shaped with two scaffold proteins wrapped around it like a belt, Shih went to Argonne National Laboratory in Illinois to conduct small-angle X-ray scattering experiments to help confirm the shape of the nanodisc. But this experimental method only gives indirect information about the actual geometry of a biomolecules like the nanodisc, generating a line on a graph, which can also be calculated through the theoretical equivalent of a small angle X-ray scattering experiment, yielding the same graph as the experimental observations. With the results from Argonne and from molecular dynamics simulations, Schulten felt very confident publishing his image of the nanodisc, which came out in a 2005 paper. Schulten and Sligar would continue to collaborate for the next four years, and in this way Schulten grew very familiar with the science behind nanodiscs.
So when Roland Beckmann showed Schulten a poster of Beckmann's newest system, Schulten instantly recognized the nanodisc: “So for me it really was a wonderful moment,” Schulten recalls. “Basically there was this disk; we were always talking about this disk, but we never really saw the disk. It was something I worked on for ten years, and I could literally see it there, with this cryo-electron density map.” The excitement for Schulten was only beginning at this point, as the system he was about to illuminate in atomic detail was filled with breathtaking biology.
Gotcha! A Protein Caught Red-handed
“It was a real challenge,” Klaus Schulten says about the new project he was about to embark on with Roland Beckmann, revisiting the nanodisc. “It was not only technically appealing, but it was also highly relevant from a scientific point of view.” Roland Beckmann had read about Sligar's nanodisc technology and realized he could use it to take his studies on the ribosome further than anyone ever had.
As discussed above, ribosomes make proteins, but a key question to consider is: Where then does the protein go? As a ribosome makes a protein, the nascent protein comes out of an exit tunnel, and at this point the protein is between thirty to forty amino acids long. Fully synthesized proteins are anywhere from fifty to thousands of units long. If an average protein chain is 200 or 300 amino acids long, then clearly some of the protein will be still in the exit tunnel and some will be outside the tunnel in the course of its assembly by the ribosome. A newly forming protein is destined for a specific place upon its completion. Some proteins will just live in the cell, in its cytosol. Other proteins may be destined for life outside the cell, and will pass through the cell membrane; for example, certain bacteria in the human gut secrete enzymes (which are a type of protein) that human cells don't normally make, but are needed to break down certain food components. These enzymes pass out of a bacterial cell. Lastly, some proteins have to make their way into the membrane of the cell, and, as mentioned previously, these are called integral membrane proteins.

James Gumbart and Klaus Schulten and the ribosome-nanodisc system they elucidated in atomic detail.
As it turns out, when proteins are secreted, or when they are membrane proteins, they sort of have a similar fate, because the first thing they see is the membrane. The very first amino acids of a protein exiting the tunnel are then usually a signal that tells the ribosome to kick in the machinery to look for the right entry point in the membrane. This entry point is itself a protein, and is a protein-conducting channel, called the translocon, and this channel either passes a protein through it, or opens a side door opens through which the nascent protein weaves its way into the membrane.
The innovation of what Roland Beckmann dreamed up was to catch a snapshot of a ribosome making a protein that was in the process of entering a protein-conducting channel sitting in an actual bit of membrane. He essentially used a nanodisc to hold the protein-conducting channel, to mimic more closely its natural membrane environment. It was thought that the structure and activity of the protein-conducting channel was very closely related to its surrounding lipid membrane. So capturing a nascent protein, still on the ribosome yet entering a membrane, with electron microscopy was quite a feat.
Beckmann worked for years on fine-tuning this system, and wanted Schulten's help in delineating the atomic detail of the electron density map using MDFF. With Schulten's years of experience already in studying the nanodiscs, this was a natural fit for him, yet he never dreamed he would return to studying nanodiscs in such a configuration. Schulten actually felt that so many aspects came together so well that it was like a gift from heaven. Work on this project was carried out by one of Schulten's star students, James Gumbart, who actually devoted his entire thesis and most of a postdoc period at the University of Illinois and at Argonne National Laboratory to the study of nascent protein membrane insertion. In 2011 the collaborators published, and Schulten relates that Steve Sligar was extremely excited when he saw the paper, as it was a breakthrough for his nanodisc technology. Schulten also feels that this illustrates the important role computers, via MDFF, have assumed in science: “This is one of those nice examples where computing becomes a key partner of experiment.”
Improving the Method: Membrane Curvature
By the end of 2007, the Schulten group had a good working model of MDFF, and was testing it against benchmark studies done by Maya Topf at Birkbeck University of London to gauge its accuracy. January 2008 marked the submission of the initial methods paper, where the group introduced MDFF and its intricacies to the scientific community. And by late 2008 the Schulten-Frank team would submit their first application paper, on the elongation factor work mentioned above. Now with a rudimentary working model, the next logical step was to start improving MDFF. Out of a totally unrelated project occurring simultaneously in the Schulten group would arise an enigma that seemed destined for illumination by MDFF, yet another in a long string of coincidences in this history; at the same time this investigation marked the next major upgrade for the burgeoning methodology.
Photosynthesis has captured Klaus Schulten's imagination since his graduate school days in the early 1970s at Harvard. While delving into the subject as a postdoc only briefly in a single publication in 1978, Schulten literally had to wait ten years to study this important process again in great detail, when the structure of a key membrane protein called the photosynthetic reaction center was first published. As mentioned above, this was the first membrane protein ever to be detailed, and warranted a Nobel Prize in 1988 for its investigators. This breakthrough, the reaction center structure, had a profound effect on Schulten. Because he was so eager to now run calculations on the published structure, he launched a preposterous plan in 1987 to build his own supercomputer, a story told in more detail here, and this would eventually lead to the parallel software called NAMD produced by his group. The reaction center structure would also reignite his research into photosynthesis, a multi-faceted journey for Schulten chronicled in this history. Suffice it to say that in 2008, when Villa and Trabuco were devising MDFF, Schulten also had a vibrant effort in his group dedicated to various aspects of photosynthesis.
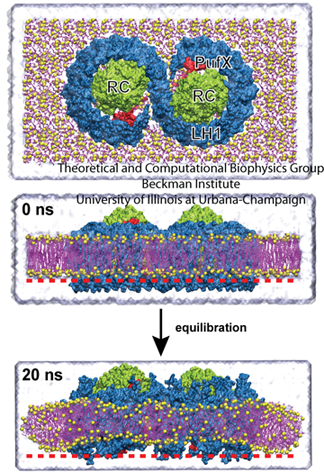
The core complex, top. The bottom image shows how the complex induces curvature in the membrane, producing a slight V-shape compared with the image at 0 ns.
One such effort involved membrane curvature. Plants are not the only organisms on earth that employ photosynthesis, as some bacteria use it as well to garner energy from the sun. In certain photosynthetic purple bacteria, a cell is populated with hundreds of chromatophores, which are compact cellular machines that perform the various steps of photosynthesis. While chromatophores are flat in some types of bacteria, they are spherical in other types. A chromatophore membrane, flat or spherical, is composed mainly of protein, with little (about 10 %) lipid content. Three key types of proteins are the reaction center, and light harvesting complexes I and II. Around 2007 Schulten had two students, namely Danielle Chandler and Jen Hsin, looking at these complexes and their curvature. “We began to wonder how the spherical membrane vesicle forms in the first place,” says Jen Hsin, with the important word being spherical, “and our hypothesis is that maybe each of the photosynthetic complexes has some sort of capability to bend the membrane slightly. And then when many photosynthetic complexes come together, a spherical membrane results as a collective effect.”
Hsin studied the core complex, which is comprised of two reaction center proteins enmeshed in light-harvesting complex I that wraps around it in an S-shape, as shown in the top image of the graphic. This structure has a third protein as well, PufX. As can be seen in the image, since there are two parts to the core complex, it is thought of as a dimer. Hsin made a model of the entire core complex from bits and pieces of the protein structures, as there was no full structural information on the complex in existence. And when she ran molecular dynamics calculations, they revealed some surprise. The core complex would not stay flat in the membrane but kept transforming into a bent, slightly V-shaped geometry, visible in the image. “We were puzzled by this result, as such geometry was unusual for integral membrane proteins,” Hsin notes, “until our collaborators informed us that they had just obtained a low-resolution electron microscopy map of the same protein complex showing that it is indeed bent–and even more drastically than we saw in the simulation.” The electron microscopy map came from Neil Hunter, who works out of the University of Sheffield.
All the ingredients were present and circulating in Schulten's group in 2008: atomistic models of the dimer, a low resolution electron microscopy map of the dimer, plus a new method, MDFF, that was suitable for combining those two types of data. It was clear that this was a situation where MDFF could prove useful.
But up until that time, MDFF had been performed in vacuo, meaning that the environment for the organism under study was neglected. But the dimeric core complex, in its natural state, lived in a membrane surrounded by aqueous solution and ions. Trabuco had to alter some code, but he figured out a way to account for the natural environment, and subsequently Jen Hsin performed MDFF for the dimer, including water, membrane, and ions. “That shows that you can do MDFF not only in vacuum, but you can add all the other medium to it and then get better results,” summarizes Schulten. “And so this was, methodologically, an interesting step, and it led to a beautiful result.” What they uncovered was why the dimers had a tendency to tilt and how that contributes to the shape of the chromatophores. “This effect doesn't just introduce membrane curvature,” Hsin adds, “but also can lead to the spontaneous aggregation of protein complexes and the formation of functional membrane structures such as membrane vesicles or tubules.”
Forged in the Heat of Battle
While the first paper detailing the intricacies of the new method, MDFF, debuted in 2008, since then Schulten has been working on many projects with Roland Beckmann and Joachim Frank and other experimentalists, and fine tuning the method as well. But the path was not always smooth. Although MDFF was catching on, a new method in science usually is subject to skepticism from various corners.
For example, in the project with Roland Beckmann and the nanodisc, where a nascent protein leaving the ribosome exit tunnel is entering a protein-conducting channel, the paper was rejected by Nature, which is the preeminent journal in the science world. “They said that we needed a chemical test, to make absolutely certain that what we suggested we saw there, particularly how the protein comes out and goes through the channel to the membrane–an essential element–needed to be pinpointed down chemically, by making chemical cross bonds between the protein that goes there and this protein channel,” recalls Schulten. He believes that the referee who rejected the paper was aware of a recent error that had just come to light in another group's publication with the ribosome and a protein-conducting channel, and thus was being overly cautious. Beckmann had worked for years developing all the elements of the system and putting all the pieces together; adding one more step, a chemical cross links test, was virtually impossible with the intricate system. “This was really the limit of what we could do,” Schulten argues. “We were climbing the highest mountain in the world, and now they wanted us to build a royal palace there.”
As a publication in Nature would have yielded almost instant attention, Schulten feels like the results, which delineate what happens to a nascent protein as it leaves the ribosome, are slowly catching on and that historically the paper, in the journal Nature Structural and Molecular Biology, will be recognized.
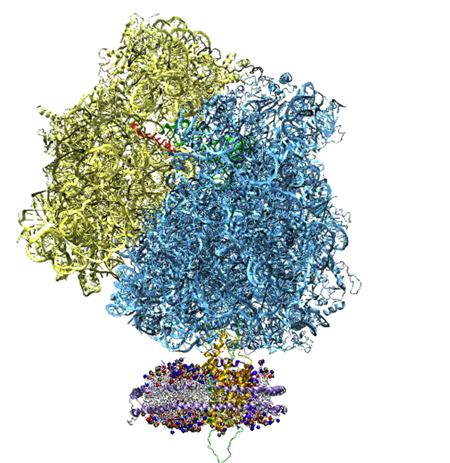
The ribosome, top, and the nanodisc, bottom.
While getting a rejection from a top journal is one thing, Schulten also was confronted in public about MDFF. He was in Colorado, at a Keystone Symposium on Molecular and Cellular Biology, and presenting MDFF, which was at the time in its infancy. He says he was viciously attacked by a famous crystallographer, who said what Schulten was trying to do was all rubbish. Schulten believes he was targeted for two reasons. First, no one likes competition. “Clearly MDFF is a new method that looks like it can get structural information that was in the past mainly within reach of pure crystallographers,” notes Schulten. “So they of course attack you then, because they believe that their methods are much better than anything else.”
The second reason he fathoms is that people believe what his group developed is not really new. To understand why Schulten disagrees with this claim, it is helpful to review the techniques that were being used before MDFF came along, to fit crystallographic data into electron microscopy maps, to put it in historical and conceptual context. In the simplest case people took the frozen structure and tried to fit it by translation and rotation into the electron density map, but as a whole, frozen form. Alternately, they took pieces of the crystallographic structure, domain by domain, and moved them independently into the map. “We say,” counters Schulten, “that the macromolecule is intrinsically flexible as can be described through a molecular dynamics simulation. Within MDFF the macromolecule can do any kind of physical motion that is consistent with what people learned about the geometrical and physical characteristics of macromolecules and had cast into the form of a molecular dynamics program.” In other words, accounting for mechanical flexibility, but only to a realistic degree, was their novel contribution.
Defending MDFF from outside attacks may be part of the usual journey for him, but regarding certain critiques, Schulten goes on the offense. He realized there was a serious argument against MDFF that he should address before anyone accused him of it. “What can happen is what you call over-fitting,” explains Schulten. “You give the system too much flexibility, and you can morph an elephant into a mouse.” It turned out that molecular dynamics was rigid enough that it did not over-morph things or over-fit things excessively. Additionally, to combat this problem, Schulten's team developed restraints to preserve structural elements and stereochemical correctness, and in fact, the development of restraints to improve MDFF is an ongoing process in the group. During the fitting procedure, the atoms are driven to high-density regions through the application of steering or driving forces, which are artificial. “Of course there is the danger that this additional force can override subtle physical properties of the model,” declares Schulten. “Particularly since you have a tendency to be impatient–you want to get results–and you want to make these forces strong so that a crystallographic structure quickly transforms into a structure matching an electron microscopy density map.” To combat errors that arise from the artificial forces, there is a parameter to control how strong the steering forces are during the procedure. Between the parameter and the restraints, users of MDFF can, if they are careful, sidestep over-fitting.
Regarding another argument detractors may have, the very first methodology paper, published in 2008 in Structure, discusses a relevant point about the atomic structure MDFF yields. In the cryo-electron density map, unless the resolution is better than 3.5 Å, it is not possible to fix the positions of the side chains, which are a constituent of the subunits of the proteins. The full atomic model coming out of MDFF is an instance; it is one of the many possible structures that are consistent with the observed electron density map. Thus, the atomic model should be understood as one representative of a larger set of structures compatible with the density map.
It is certainly ironic that although Joachim Frank waited years for Schulten to finally collaborate, Schulten still got into the field almost at its inception. And this early entrance has played a key role in the success of MDFF. Combining structural information from different sources is collectively referred to as “hybrid methods.” Right now many scientists are formulating their own hybrid methods, which Schulten recognizes will compete with MDFF. The group that Schulten heads is theoretical and computational, but target audiences for MDFF are structural biologists, i.e. experimentalists. New users of MDFF need to develop some feel for molecular dynamics simulations, as well as learn NAMD and VMD, which are barriers that might be high for some experimentalists. One of the pioneers of MDFF, Elizabeth Villa, now working on a new project in a group in Munich, still moonlights in MDFF, helping various experimentalists from time to time run simulations. She has a unique perspective, as she understands the inner workings of MDFF and is still in the trenches helping people use it. “We were lucky that it worked really well, had a lot of visibility, and we also caught the market early,” Villa remarks; “To come and release MDFF right now, as fantastic as it is, would never have the impact that it did at the time.” Villa also, like Schulten, is aware that new hybrid methods are emerging all the time–MDFF competitors–and that some work relatively well.
Competition is something Schulten fully understands from working in science for over four decades. “You can never lean back, you always have to be sitting on the edge and watching out,” remarks Schulten. “There is always something new, and new young and energetic people are joining the race every academic year.” But Schulten's group is marching forward too, working on improving the method and branching out to apply MDFF so that it is more than just a hybrid method for cryo-electron microscopy and crystallography, developments covered in the next half of the story.
While Schulten understands that simpler methods are starting to crop up, and that interested users will consider both quality and ease of use when opting for a new hybrid method, he is proud of the impact MDFF has made in only five years. He points to the large number of papers published by groups outside his, some in prestigious journals like Nature and Science. (A complete list of these papers is included in an appendix at the end of this article.) “Usually it takes ten or twenty years before people are really using somebody else's method, but here we had a very quick adoption,” Schulten explains. “It doesn't happen every day, I can tell you that.”
The Advantages of Youth
“I have this competitor, David Shaw, a great person, gentleman and a billionaire. He puts several hundred million dollars into macromolecular modeling, like what we do here. How can I compete with him?” Schulten poses a good question, but it is one that he has an answer to. He has realized that his young people are the capital that permits him to even vie with billionaires. Most are eager to learn, eager to prove themselves, and some dream of being great in science. Schulten recognizes as well that his graduate students and postdocs, who often fall between twenty and thirty years old, have fewer family responsibilities and fewer health problems.
Klaus Schulten surrounded by some of the young talent who helped shape MDFF. Leo Trabuco on his right, and J.C. Gumbart on his left.
Schulten points to the archetype of the professors who relentlessly siphon off all the energy from their young people. While he jokingly wonders if some see him like this, he feels he offers something in return. “The most important thing I can do as a professor is give somebody a good research problem,” Schulten counters. “A good research project, that is the hardest thing to find, but that is actually the best thing you can give to a young person.” He feels that he gives them a direction, so that they apply themselves to something good for themselves, both career-wise and as people. “When I was 22, I was basically sitting in front of a blank sheet of paper, wanting to do biological physics, but I didn't know what to do,” remarks Schulten. “I was full of energy and my head was exploding. I knew what I wanted, but I didn't know where to start.” His advisor gave him a good project to work on, and he did the rest himself, but he was very grateful for the gift.
Schulten, a seasoned scientist who has spent the last forty years always trying to break new ground, realizes there are parallels between what he is trying to do in science and the rest of life. “You build a totally new house; you build a new company from where there was nothing; you develop a new scientific field. There is an ENORMOUS amount of work involved in doing something really new,” Schulten explains, with the caveat that he cannot express adequately with words the effort this takes. In building up a new method, like MDFF, necessarily a lot of time is used up in and learning from errors going in the wrong direction since it is not clear what exactly to do, and then there is assembling the bases to make a foundation on which to create the edifice and the knowledge. “And that, only young people can do, because it involves so much energy, not only simply intelligence and ability, but also, just physical energy,” Schulten concludes. The pioneers of MDFF were certainly energetic graduate students, but now, in the next stages of MDFF, Schulten has assembled another team of energetic young researchers, whom we will meet in the following sections. They are poised to refine MDFF and utilize it for many scientifically relevant issues.
The Advantages of a Veteran
I asked Klaus Schulten if he saw any advantages or disadvantages to starting a new field so late in his career. In 2007, when MDFF really began, he was 60 years old. He replied by offering a joke.
A Catholic priest, a Protestant reverend, and a Jewish rabbi sit down to discuss the controversial topic of when life begins. The priest says life starts at the moment of conception, and then the reverend says perhaps at two or three months. They turn to ask the rabbi the same thing. “I believe much later than that,” says the rabbi. But when exactly, the others want to know. “Oh, when the kids leave home and the dog is dead.”
Schulten says this reflects his situation accurately: “We have a daughter who is out of the house. My wife is also a professor so she is not expecting that I come home for dinner punctually at night. And our dog is long gone.” While he realizes he may have only one tenth the energy of a young faculty person, he has about ten times more time these days. Of course, he also works long hours and weekends, but can do that with his lightened family responsibilities.
He feels like the productivity of his group in the last ten years also reflects the efforts of a much younger scientist, with its many software contributions as well as the new science unveiled. Schulten has at least ten active areas of research, most vastly different from each other, and on top of that he supervises the group's two software products, NAMD and VMD. It is really amazing that he had time at all to learn a whole new field, i.e. hybrid methods, and on top of that he needed to come up to speed on the ribosome. He offers two more reasons why he's been so productive at the latter stages of his career. First, his health has been good. And second, his group at the Beckman Institute was established in 1989 and by now is a well-oiled machine with little fluctuation in staff.
The confluence of factors all came together perfectly for the creation of MDFF. Klaus Schulten's career was ready for it, in fact.
But when contemplating if he should take on new projects, Schulten realizes he has limitations this late in his career. “Every big thing you do in science takes about ten years,” explains Schulten. “For something to come to real fruition, it takes time.” With retirement coming, Schulten says he now has to be careful when considering potential prospects because he might not be able to do a project justice if he can only work on it for a few years intensely. In this way, the timing of MDFF was perfect for Schulten; he started it when he could foresee at least ten years to devote to it. That it has gained widespread appeal before Schulten retired is lagniappe, as he figures MDFF was possibly the last big thing he could afford to take on.
“So it was not a problem at all to do this late in the career because the circumstances were totally ready for it.” Very simply, Schulten's main goal in his professional life has been to understand living cells. But a cell is a complex entity that involves the assembly and cooperation of many macromolecular systems. Since Schulten wanted to understand living cells through theory as opposed to through experiment, he needed tools to simulate and visualize large systems, which led to the creation of NAMD and VMD. When Joachim Frank came to Schulten with electron microscopy images of a molecular machine and its various components, Schulten immediately recognized electron microscopes capture large biological systems, an appealing prospect, and that characterizing such systems was in keeping with his long-cherished goal of describing biological organization. And he had the software in-house to meet just such a challenge. “So this was for me a beautiful opportunity to cap off my career,” reflects Schulten.
While MDFF fit really well into Klaus Schulten's life, and appeared not a minute too soon, the genesis of the method really is a combination of a veteran with ample time and a crew of young talent with ample energy. And for the next phase in MDFF's young life, Schulten has assembled more raw talent, a cast of characters we will meet below. He has also found himself amidst a flurry of exciting applications and is charting new directions for the methodology as well.
That Touch of a Developer
By 2010, both students who pioneered the method had graduated with PhDs from Schulten's group, and Schulten had other graduate students step in to continue working with MDFF, but to him it became clear that the methodology needed systematic development. And what that meant to Schulten, whose group develops software, is that he needed to get a computer programmer involved. This required finding a source of funding, however. In the great lattice of coincidence, when Schulten looked into it, he found that the NIH had funds available for a software developer in the field of hybrid methods for structure analysis, an exact match. And when they advertised for the position once the funding was secured, Schulten was thrilled to find another exact match.

Ryan McGreevy, the main software developer for the MDFF project.
When Ryan McGreevy was an undergraduate at St. Mary's College in Maryland, he couldn't decide what he liked better–biochemistry or computer science–so he decided to double major in both. Upon graduation he worked for about a year doing military flight simulations as a systems engineer. “I missed the science aspect,” McGreevy said about his first job, “because I also worked in labs throughout college. And then to go from doing a lot of science to none at all, it was kind of jarring.” He also enjoyed the investigative aspects of science and the constant process of learning new things. So it is easy to imagine what a great match it was for him to find the MDFF position, which he started in August 2010.
“As I have a background in biochemistry, I fit well into the group since I can speak the language of the scientists,” says McGreevy. The Theoretical and Computational Biophysics Group in Beckman is unique in that it is a mixture of software engineers and research scientists, and interaction between the two camps is essential for smooth functioning. Schulten wants his software developers to have respect as well as a mindset for science, to facilitate communication and understanding. While McGreevy did not specifically have a background in biophysics, his training in biochemistry was still invaluable. “I knew enough of the vocabulary and what was going on that I could almost immediately start talking with the scientists and figure out what it was that they wanted to do, and what they were doing, and why things were being done the way they were,” offers McGreevy. He was able to get a running start.
His role on the project was clear: bring a developer's perspective to MDFF. While the original code and plugins written by the pioneers worked well and produced many early successes, there were rough edges. “As soon as you try to adapt the old features to new systems, or use these features on larger, more complex structures, or if you want to expand your tool set and introduce some new functionality, going back into that old code can be very difficult,” says McGreevy. Some of the things he now does are fix old code and make it cleaner and more efficient–as well as easier to use. And McGreevy enjoys the challenges of the project, and the exposure to so many new concepts. “It really allows me to flex all of my intellectual muscles, learning about all this stuff and using all this information that goes into this method,” he reiterates.
Obviously he has had to teach himself a good bit of science to function well at his job, and the code development aspect of his work benefits from being in a group with software engineers. He can talk to John Stone or Jim Phillips, senior developers of VMD and NAMD, respectively, for advice and tips on how to make his code highly efficient. He takes full advantage of the resources available in Schulten's large group. And this is making MDFF a better product in an atmosphere of increasing competition.
Further Improvements
While Kwok-Yan Chan may have majored in physics as an undergraduate in China, he is curious about all aspects of science. He was fascinated by genes and how they turn into proteins, and so decided he wanted to slightly shift fields and study biophysics in graduate school. “Imagine, like you or me, our genes are probably ninety-nine percent the same, maybe one percent or less is different, but how can that make such differences in us,” wonders Chan.

Kwok-Yan Chan, a graduate student working on MDFF.
When he started graduate school at the University of Illinois in 2009, he went to talk to Klaus Schulten and asked if he could work on DNA and genes. But Schulten told him what Chan wanted to do was solved already; however, Schulten had an alternative idea: work on the ribosome, which is how DNA turns into messenger RNA, then proteins, and join the MDFF team. After Chan read an MDFF paper, he was fascinated and by that time, hooked. He now spends about half his time doing applications, mainly with the ribosome, and the rest in methodological development.
One of his first projects was to try a new method to reduce the computational resources needed in MDFF, since it often is applied to massive systems. When one simulates proteins in a natural environment, often a water box is used to surround the protein. This increases the number of atoms to be simulated by ninefold or tenfold. Another graduate student in Schulten's group, David Tanner, had just been working on a new feature of NAMD called the implicit solvent method. It is a way to account for many of the physical characteristics of the water box, but one treats it actually as a kind of continuum background; Tanner implemented it the method parallel. Chan took the new feature in NAMD and tested it with MDFF. He discovered that it still gave good models but reduced the computational demands greatly.
At this point in time, Ryan McGreevy was hired as the new MDFF developer, and he and Chan joined forces to add some new restraints to enhance the method. As mentioned before, MDFF works by adding a steering force to guide atoms to the electron microscopy density. It can be difficult to give the system enough push to get it to go to the electron density map, while also keeping it structurally intact everywhere–a large steering force could pull it apart. There were restraints already in place to keep protein secondary structures intact, to prevent unwinding helices or pulling apart beta sheets. But then Schulten, Chan, and McGreevy realized they could define a type of higher level of restraint, that is, pick any arbitrary set of atoms, define that set as a “domain”, and keep it together. “Suppose you have some domain of a protein structure, and you know that it is correct, relative to itself, but it just might not be in the right position,” McGreevy explains. “So you're moving the whole piece, but the parts of it are where they need to be relative to one another, so you want to keep them together as you move it.” For example, McGreevy says this new restraint was used on the two large portions of the ribosome (each one considered a domain) that perform the ratcheting motion Joachim Frank first described in 2000.
Symmetry restraints were applied to the helical subunits in red, while the gray subunits were fitted without them, with edge effects present. Image of a bacterial nitrilase.
Biological macromolecules often have symmetrical relationships between the subunits of their structures; for example, virus capsids are often made of symmetric subunits. There could be hundreds of the same subunit repeated–they are all the same, just rotated and translated through space to give an overall tube or cylinder or sphere. MDFF can take advantage of this.
“For the symmetry restraints, you define these subunits to be all symmetric to one another,” says McGreevy about the new feature. “So you sort of combine all the symmetric subunits and get the average structure that represents them. And then you restrain them all to that average structure.” Basically each subunit has to be the same, as in these cases the structures are made from identical twins or triplets and so on. An example where accounting for symmetry is helpful is when the electron microscopy densities are not uniform even though the underlying molecular subunits are identical; areas can be washed out due to motion or disorder, and thus be of much lower resolution than other areas. But if it is clear that the structure in the washed-out area is made of symmetric subunits, they will be kept in the same shape as the subunits in the better-defined regions of the map. Another example of the use of symmetry restraints is when a subunit is fitted into a region of larger density. In the simulation, it is possible to have edge effects, where the density next door can pull the edges of the subunit and errors result; with symmetry restraints, these edge distortions can be avoided.
MDFF and the GPU
In Klaus Schulten's group, simulation of large systems is a major aspiration, and his software developers have to constantly work to make their products fast and efficient enough to handle systems that are only becoming bigger and bigger. One technology that has been incorporated into NAMD and VMD is the graphics processing unit, or GPU. In its most simple form, the GPU, as its name implies, is a chip used to boost graphics performance on a computer–for example, to render more realistic scenes in computer gaming. Shortly after their advent at the turn of the new millennium, scientists realized that GPU's unique features could accelerate the calculations they were doing, and that they were especially useful in parallel computing. Both NAMD and VMD have taken advantage of GPUs to an ever-increasing degree in the last ten years.
Presently Ryan McGreevy accelerates MDFF calculations through the use of GPUs. The core of MDFF is the tool Gridforces, which, as mentioned previously, makes a 3D grid of forces from the cryo-electron density map. These Gridforces computations are usually intensive and can hurt overall speed. “It just so happens that the calculations involved in figuring out the Gridforce potentials are actually very good candidates to be parallelized on a GPU,” McGreevy says. “There's actually special hardware in a GPU that does exactly the needed calculation VERY quickly.”
The developers of MDFF are working on providing instant feedback about the quality of fit by harnessing GPUs.
A natural first step utilizing GPUs for the Gridforce feature is to simplify the overall organization of the needed calculations. Since this feature was designed by Alek Aksimentiev and David Wells to be very general, it has aspects MDFF does not need, and it can slow down the computations or add unneeded complexity. David Wells, with input from McGreevy, has trimmed down Gridforces so that it performs just the calculations relevant to MDFF. The outcome is something they've named Gridforces Lite, and it is now available in NAMD. After that McGreevy created a working model of Gridforces Lite using GPUs. “The speed isn't satisfactory yet, so there's a lot of work that still needs to be done to make Gridforces much more efficient, but a prototype does exist.”
There are some analysis tools employed for MDFF in VMD that can also benefit from GPU acceleration. For example, once MDFF is run and a model is generated, it is possible to do a calculation to see how good the match between the measured electron density map and the suggested model is. For this purpose, MDFF can essentially go backwards and generate from the model a calculated electron density map. Then VMD can compare the calculated and the measured (the one from the electron microscope) electron density maps and "cross correlate" them to see how well they match. It is even possible to calculate cross-correlation coefficients by local domain, and color a model appropriately to see which sections are matched well and which ones are matched poorly. However, calculation of an electron density map from a model is very demanding and, therefore, the procedure can benefit from GPUs.
In fact, not only can cross-correlation be accelerated by GPUs, but McGreevy can tap into other GPU acceleration solutions developed in Schulten's group. For VMD developer John Stone, it has always been a dream of his to represent surfaces of molecules in the course of a molecular dynamics trajectory run. As the run progresses, the surface dynamically changes, and because rendering surfaces graphically is such a computationally demanding endeavor, Stone had to wait years to offer this calculation as an interactive option for users. But as soon as Stone realized GPUs were sophisticated enough to work on this challenging venture, the Schulten group developed a feature of VMD called QuickSurf, which debuted in 2012. If users of QuickSurf have a GPU in their computer, they can speed up surface-rendering a hundredfold, and seamlessly watch a movie of a molecular surface evolving, something unimaginable before.
“Well it turns out that QuickSurf is very similar to what we want to do when we create a simulated density,” says McGreevy. To utilize the speed benefits of QuickSurf, McGreevy then teamed up with John Stone to implement faster calculations. The two were able to see speed increases of up to 137 times the existing sequential CPU implementation when they employed GPUs for cross correlation.
Tackling HIV
One day in 2011 Klaus Schulten got a call from a long-time friend, who informed him she had the perfect project on which they could collaborate. Schulten listened eagerly, as he had been trying to find a project to work on with Angela Gronenborn since the 1980s. Schulten and Gronenborn met while both lived in Munich, Schulten at the Technical University of Munich and Gronenborn at the Max Planck Institute of Biochemistry, where she was leading a Nuclear Magnetic Resonance (NMR) group. They became fast friends, one reason being they both come from the same area in Germany; they also very much respected each other's work. Gronenborn, like Schulten, would leave Germany, finally settling at the University of Pittsburgh, where she now heads the Department of Structural Biology.

Juan R. Perilla, who simulates the capsid of HIV-1.
What Schulten heard on the phone that day was very intriguing, as Gronenborn described one of the exciting projects she had been exploring with her co-collaborator there Peijun Zhang. Gronenborn said she needed someone to merge data from different sources (namely crystallography and NMR with electron microscopy maps), and that the system in question was extremely large. As if this wasn't enough to reel in Schulten, the topic was a hot and relevant scientific system: the HIV-1 capsid.
While all the parameters fit nicely with the skillsets in Schulten's group, he knew he needed to find a “warm body,” as he put it, who wasn't too busy to take on the capsid. And he was very fortunate that he asked new postdoc Juan Perilla if the capsid was something that would interest him, as he was not yet loaded with projects. Perilla had just come from graduate school at Johns Hopkins, and as Schulten sees it, Perilla caught fire immediately with MDFF of the capsid.
But what exactly is a capsid? Well, a virus is simply a particle that has some genetic material enclosed in a shell made of proteins; this shell is the capsid. On some viruses there may also be an envelope surrounding the capsid, which is the case with HIV, although this story deals with the capsid only. The capsid protects two identical strands of RNA (along with some other proteins), and HIV is a type of retrovirus, so named because of the way the virus replicates, moving backwards in the sense that its RNA turns into DNA. This retrovirus has touched almost every corner of the globe, as UNAIDS estimates that 34 million people were infected with HIV worldwide in 2011, and at least 1.7 million died from AIDS-related illnesses that year alone. The treatments, antiretroviral drugs, are helping those who are HIV positive live longer, but as the virus is prone to mutations, patients are likely to develop drug resistance. (However, this drug resistance is only one of the many factors that make the virus so insidious; many treatises on HIV, in books and articles, as well as online, exist which can describe in greater detail why it is so pernicious.) Suffice it to say, it is a global imperative to find new treatment options.

Electron Microscopy image of the special HIV capsid developed by the Pittsburgh Center for HIV Protein Interactions.
Many scientists now work on various aspects of the virus and its lifecycle to broaden the arsenal of weapons used against HIV. And one means of attack is to look at the capsid. “Just to give you the pharmacological angle,” begins Schulten, “it is interesting to note that certain monkeys don't get HIV. Apparently they can attack the capsid on its way from first entering in the cell, to going to the cell nucleus. They attack it and interfere with its normal uncoating, and thereby they avoid infection.” To do something similar, scientists would need to know the exact details of the capsid's chemical nature, down to the atoms and atomic bonds that make up the molecules that form the capsid; accordingly, the chemistry of the capsid needs elucidation. “You need an atomic-level structure, not just overall shape, or a simplified picture. You need the detail,” concludes Schulten. “Then you see where you can attack.” MDFF is a technique that yields just such atomic-level detail.
However, the capsid is an extremely difficult object for study, and Schulten says this is really an understatement. Basically, not all capsids are created equal. There is great variety in the shape of the capsid from virus to virus. For creating a three-dimensional image of an object using an electron microscope, as we learned earlier, it is essential to have multiple identical copies of an object, a characteristic that made the ribosome so attractive to Joachim Frank. In lieu of having an ideal set of circumstances for capsid analysis, scientists still have made headway. Low-resolution data revealed that the capsid was conical. People then started to think about the components of the cone, and drew on early understandings of virus symmetries. For example, in 1956 Watson and Crick proposed that viruses are made up of identical subunits arranged symmetrically, and since then others have explored how capsid geometry (of all viruses) is dependent on the ordered arrangement of a few identical proteins. The HIV capsid was no exception, as people realized that the cone was made of protein-containing hexamers and pentamers. (This became known as the fullerene cone model, which is a geometrical approximation to the cones observed under the microscope.) Mark Yeager, working with Barbie Ganser-Pornillos and Owen Pornillos, eventually solved the crystal structure of the hexamers, although in a flat lattice as opposed to in their native curvature, and with hexamers separated from each other, rather than in close contact, as in the actual capsid.
Meanwhile, Angela Gronenborn had been leading the Pittsburg Center for HIV Protein Interactions, and making great strides in structure determination of HIV-related proteins. The center also studies, more generally, the early stages of HIV infection, and the capsid falls into this category. Center researchers, after years of tedious effort, succeeded in creating a special form of the capsid that is very regular; in this form they are essentially tubes as the image at the right shows, open at both ends like a straw, and formed of the hexamers mentioned above. With the capability of creating tubes all of one size now realized, cryo-electron microscopy could be used on the structure of the hexameric capsid.
With all the components now in hand, it seems logical why Gronenborn asked Schulten to collaborate, as she told him that he would have to deal with a huge system of an entire capsid, electron microscopy maps, and X-ray and NMR structures of the component parts. Gronenborn knew what Schulten's group worked on, as she is also on his NIH center advisory board, and was aware of his new method, MDFF, as well as of his ability to simulate structures larger than anybody else could. And Schulten was thrilled when his new postdoc, Juan Perilla, seemed interested in the capsid enigma.

The capsid proteins, with the N-terminal domain in blue and the C-terminal domain in orange.
Luckily Perilla was extremely interested and extremely capable of doing the job, as MDFF of the capsid presented many challenges. Because assembly of the special HIV capsid in vitro requires huge salt concentrations, this had an effect on the molecular dynamics run Perilla had to do initially to equilibrate the structure, a task that did prove quite complicated. Some regions of the capsid had no X-ray or NMR structure to fit into the electron microscopy map, so Perilla had to build them using homology modeling. “We had to be very careful that those regions we modeled were not going to assume faulty conformations when we applied MDFF,” Perilla explains. He also had to consider computer efficiency for such a large structure. To fit over 70 hexamers into the electron microscopy map one after another would have taken more than two weeks; Perilla got the process down to a few hours on the computer, with scripts of his own that did the tasks in parallel. In general, putting all the pieces together resulted in a simulation of over 13 million atoms, including water and ions. This was a great test of the mettle in Klaus Schulten's group.
The capsid research revealed many things. In previous work done by Gronenborn at the Pittsburgh center, her team uncovered a special dimer. As illustrated, each capsid protein has two domains, an N- and a C-terminal domain, located closer to the inside or the outside of the capsid, respectively. Each hexamer accordingly has six N-terminal domains on the inside and six C-terminal domains on the outside. Gronenborn and her coworkers showed in 2009 by means of so-called NMR structure analysis that neighboring hexamers in the capsid are held together through interactions between the C-terminal domains of the capsid proteins. In fact, the C-terminal domains cut off from their associated N-terminal domains were seen to form dimers. In the new work MDFF again revealed such dimers, but now a family of three were found; they were closely related structurally, but not identical. Present were not only the original dimer, but also two others. Since the capsid is so symmetric, made of repeating hexamers, it also exhibits a three-fold symmetry axis, a promising pharmacological target.
Getting to know the arrangement of dimers in the capsid was a key step towards revealing the atomic level structure of the HIV-1 capsid, but other key structural elements of the capsid were not known yet, namely the ones involving a pentamer of capsid proteins being surrounded by five hexamers. In this case no structural observation could be achieved and the project had to rely on computation alone. This worried the collaborators, but the needed simulations turned out such an eminently natural result that the worries turned out to be unfounded. Perilla and Schulten started out with a hexamer surrounded by six hexamers as seen in the tubular capsid, but eliminating not only one outside hexamer but also one of the six capsid proteins in the central hexamer, arranging the resulting pentamer-of-hexamers as symmetrically as possible in a flat geometry. The resulting system left gaps between capsid proteins that were closely interacting in the tubular capsid case. The investigators hoped that the gaps would close in the simulations, and indeed they did so beautifully. The simulation started out flat, but after 700 nanoseconds, the pentamer-of-hexamers segment actually curved like a dome, shown at right, the angles of the curve very close to the fullerene model. The simulation revealed that the three-fold symmetry axis was more compact in the pentamer-of-hexamer case than in the hexamer-of-hexamer case.
The hexamer-of-hexamers outlined in yellow, and the pentamer-of-hexamers outlined in blue. The two systems are shown here schematically as a fullerene model, but were actually simulated as all-atom protein systems.
At this point the investigators had both of the needed structural elements, the hexamer-of-hexamers and the pentamer-of-hexamers, available to build a model of a typical HIV-1 capsid. Here experimentalists and theoreticians could again exchange data as the experimentalists succeeded in taking electron microscopy snapshots of capsids, employing the microscope in so-called tomography mode. In this mode the electron microscope works much like magnetic resonance imaging (MRI) works in the hospital on patients, giving an image of the shape of the capsid. The collaborators sought and found a great match between a fullerene cone composed of 216 hexamers and 12 pentamers that proved to be a very close match to the electron microscopy tomogram. Perilla and Schulten pieced the model together from the now available data and subjected it to the largest ever (as of 2013) molecular dynamics simulation. The total atom count of capsid, water, and ions was over 64 million. The model proved stable in a 200 nanosecond simulation and yielded the most detailed information on the chemical structure of the capsid a pharmacologist or HIV biologist could wish for.
Overall, MDFF allowed a unique insight into the HIV capsid. It illuminated the lattice, and how the hexamers come together. Further simulations then modeled how the pentamer actually comes together with the hexamers, and biochemical data that was part of the study actually supports what Perilla and Schulten found in the more compact three-fold symmetry axis of the pentamers. “So all the building blocks, all the interactions that keep a capsid together, were pretty much unknown before we actually built this model for HIV,” summarizes Perilla. More details about the simulations, which marked a milestone in the Theoretical and Computational Biophysics Group, can be found in this video that includes interviews with Schulten and Perilla as well as explanatory animations. For the combined efforts of the experimentalists, teams from Pittsburgh and Vanderbilt, with the theorists in Urbana, this research on the full chemical detail of the HIV capsid merited a 2013 publication in Nature.
The next step is to use the results of this MDFF work, which provided the entire structure of the lattice, to assist pharmacologists in targeting the capsid for treatments of HIV infection and to reveal in the context of basic biology research how the capsid provides both a stable shell protecting the viral RNA and, in contrast, a quickly opening shell releasing the RNA after a capsid finds itself inside a host cell upon infection.
Demystifying Antibiotic Resistance
While one application of MDFF was on the HIV capsid, Schulten and his graduate student, Yanxin Liu, worked with a team of experimentalists to unearth the capsid of the rabbit hemorrhagic virus, possibly providing chemical detail helpful in making a vaccine. It is thus clear that MDFF is indirectly useful in the fight against infectious diseases. In 1994 Laurie Garrett published a groundbreaking book, The Coming Plague, laying out with copious evidence how newly emerging infectious diseases were racing ahead of scientists' ability to fight them, and that old pathogens as well were making a comeback. Part of the reason for the comeback is that bacterial infectious agents, treated by antibiotics since the 1920s, were developing drug resistances. Today certain bacteria, such as Staphylococcus aureus and certain strains of tuberculosis, are almost completely resistant to even the most powerful antibiotics.
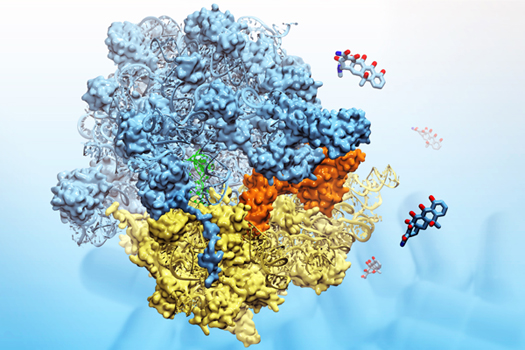
The E. coli ribosome bound to the orange-shaded Tet(O), a protein responsible for tetracycline resistance. Molecules of the antibiotic are in the background.
In a climate where the microbes are outsmarting the scientists, it is important now to understand the mechanism of antibiotic resistance. One small step in that direction comes from an application of MDFF that Schulten's group did with Joachim Frank, the man who motivated the pioneers to create MDFF in the first place. The topic at hand is tetracycline resistance, and again features a key player, the ribosome.
Frank and his group succeeded in gaining a very high-resolution map of a subunit of the ribosome with a protein bound to it that is responsible for the drug resistance of tetracycline, a popular antibiotic. Earlier results of the ribosome-protein complex were only documented with fairly low-resolution maps. Yan Chan from Schulten's group (featured earlier) was tasked with fitting the pieces into the electron density map, and he explains how tetracycline is meant to work: “These antibiotics, when they are working well, bind to the ribosome [of the bacteria it is meant to kill]. And it binds to the place where the next transfer RNA comes in. But because its place in the ribosome is being occupied, the tRNA cannot come in, the bacteria cannot make proteins for themselves, and so they die.” (Just for reference, tRNA brings to the ribosome the amino acid that will be the next element of a nascent protein.) One path to drug resistance happens when the target bacteria manufacture so-called ribosome protection proteins; these proteins go to the site on the ribosome where the antibiotic has inserted itself, and free up the site so the ribosome can start making its proteins again. The ribosome protection protein in question in this study is called Tet(O).
Part of Chan's task in running MDFF for Tet(O) bound to the ribosome was to use homology modeling to create a working, atomic-level structure of this protection protein to fit into the map. It was critical to see the conformational changes in the ribosome and the exact behavior of Tet(O) as it was attached to its binding site in the ribosome. MDFF revealed that when Tet(O) binds to the ribosome, there are three loops of functional importance, which are critical when the protection protein interacts with the host binding site. Other experimental collaborators on this project then did mutation studies on Tet(O), making the protein shorter or removing some of the loops. When the mutations changed the loops in specific ways, the bacteria no longer had resistance. The mutation studies validated, thereby, the structure given by MDFF and underscored the importance of the loops that MDFF was able to capture. Through MDFF, scientists could see the mechanism of a particular type of antibiotic resistance at the molecular level.
Conclusion
While MDFF has had fabulous successes in its short life so far, Schulten has many new directions going into the future, both methodological developments as well as exciting applications. He is certainly not sitting still.
As development director for two popular products, VMD and NAMD, it is critical for Schulten to have a sophisticated user interface on all software associated with his group, in particular also the MDFF software. The developer hired in 2010, Ryan McGreevy, is working on a graphical interface for MDFF. Right now there are many tools in VMD that users have to manipulate when using MDFF, but McGreevy is trying to bring them all together into a single front end for all the tools; users would have check boxes with options instead of always typing in commands. “But because there are so many tools involved in MDFF, it takes some effort to use them correctly, and we need to develop a front end such that it IS actually helpful to the user,” he states. McGreevy, an excellent communicator himself, luckily can interact with the many users of MDFF in Schulten's group to develop this software interface in the most user-friendly way possible. In the current climate, where new hybrid methods are rapidly emerging, ease of use will certainly be a factor when structural biologists consider which fitting method best suits their needs.
So far, MDFF is a technique to coax X-ray crystallography structures into lower resolution electron microscopy maps. But Schulten has in the works an offshoot that would fit high-resolution X-ray data or computationally predicted structures into poorer-resolution X-ray data. There are many uses for the new MDFF. For example, in a pharmacological laboratory, scientists might be interested in seeing how different molecules, potential drug candidates, bind to certain receptors on the surface of a cell. The first time they crystallize the drug-receptor complex, they might get a high-resolution structure. But then in trying new candidates, they might work out lower resolution X-ray diffraction data. Schulten imagines that in such a situation scientists will take advantage of this upcoming new method, xMDFF. This might also be of interest to researchers who use high-throughput crystallography methods; they can generate structures rather quickly but the data is not as good as the more traditional, slower techniques. “So our goal is to have a comprehensive set of tools which can take low-resolution crystal data and refine it to a much-improved structure from what you had to begin with,” summarizes McGreevy.

The HIV-1 capsid, a 64 million atom structure simulated with petascale supercomputers.
Suppose a scientist generates an electron microscopy map of a macromolecule, but no crystal structures exist to use for fitting, or perhaps only parts of a structure are available. Schulten has this scenario covered. The answer: use a computer to generate an atomic-level structure as a starting point for MDFF. Today computer structure prediction has become very successful, and, not to mention, it is efficient and cost-effective when compared to a laboratory operation. For this endeavor, Schulten has teamed up with Dong Xu, a former student of his in the early 1990s, who is now a professor at the University of Missouri. Schulten's team is tapping into Xu's brainchild, MUFOLD, a protein structure prediction program. Schulten and Xu and their respective crews are working on a product to be called MUFOLD-MDFF. “This might likely be what everyone is using in the future because this method is becoming better and better,” Schulten says. “You might not even need crystallographic structures anymore. You would just take the predicted structure to use for the fitting.”
While Schulten is charting many new paths for MDFF, he is also pushing the method to its limits with exciting applications. As mentioned above, Schulten and Perilla are doing further compelling research with the structure of the HIV capsid they obtained from MDFF. And Schulten's group continues its studies on the ribosome and uncovering the fascinating biology on proteins related to the ribosome. Currently they have a project focusing on a newly-forming protein in the ribosome, and the protein called the “trigger factor” that chaperones it, once the nascent protein gets outside the ribosome exit tunnel. This project is so intensive that Schulten needs all the power of the next generation of supercomputers, the petascale computers that assisted the group already in solving the structure of the HIV-1 capsid. This will be a big test for the method, but should yield worthwhile results. Schulten team members, in the short time that MDFF has been ongoing, have already produced numerous structures in atomic detail for other scientists to use; those structures are on view in a database called the Protein Data Bank, with the contributions by Schulten's group found here.
MDFF started from humble beginnings, with Schulten and his graduate students motivated by a fascinating molecular machine imaged by Joachim Frank. But it has turned into a very successful method in only a short time, a phenomenon Schulten says he rarely witnesses in science. Part of the success of MDFF is based on its effectiveness, plus the early entry on the scene, but there is another more intangible aspect as well. Klaus Schulten is the kind of scientist who wants to share. He produces two free software products for the scientific community, and is willing to put their latest advances immediately in the hands of his competitors. He tells stories of meeting scientists at conferences who confide they would never want to hand over their hard-earned methods. Not only does Klaus Schulten believe in making his methods open and available, he has years of experience doing it. Whether it is dissemination, workshops, or online tutorials, Schulten's team has the relevant experience. Schulten himself has trained face-to-face over a thousand people on NAMD and VMD. MDFF is probably so successful because its genesis was in a large group in which all the elements for its creation and subsequent expansion were present. Moving forward on MDFF, with a talented crew of young scientists surrounded by a treasure of resources, veteran Klaus Schulten can afford to dream expansively.
Other Related Articles
If you enjoyed this article, you might be interested in these other histories:History of Magnetoreception
History of Polyenes
History of Membrane Proteins
History of Bionanotechnology
History of Photosynthesis
History of NAMD
History of VMD
Comments?
For comments or inquiries, Lisa Pollack can be reached at the following: lpollack AT ks.uiuc.eduCitation
To cite this article, please use the following:Pollack, Lisa. "Evolution of a Method: Historical Perspective on Molecular Dynamics Flexible Fitting." 30 May 2013, from http://www.ks.uiuc.edu/History/MDFF/
References
Joachim Frank. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. San Diego, Academic Press, 1996.
Joachim Frank. Single-particle reconstruction of biological macromolecules in electron microscopy–30 years. Quarterly Reviews of Biophysics, 42:3, 2009.
Joachim Frank and Rajendra K. Agrawal. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature, 406:318-322, 2000.
Emily F. Freed, Franziska Bleichert, Laura M. Dutca, and Susan J. Baserga. When ribosomes go bad: diseases of ribosome biogenesis. Molecular BioSystems, 6(3):481-493, 2010.
Joachim Frank. Three-Dimensional Electron Microscopy of Macromolecular Assemblies. New York, Oxford University Press, 2006.
Joachim Frank. FOUND IN TRANSLATION: Collection of Selected Papers with Commentaries. Singapore, World Scientific Press, in press.
Elizabeth Villa, Jayati Sengupta, Leonardo G. Trabuco, Jamie LeBarron, William T. Baxter, Tanvir R. Shaikh, Robert A. Grassucci, Poul Nissen, Måns Ehrenberg, Klaus Schulten, and Joachim Frank. Ribosome-induced changes in elongation factor Tu conformation control GTP hydrolysis. Proc. Natl. Acad. Sci. USA, 106:1063-1068, 2009.
Birgit Seidelt, C. Axel Innis, Daniel N. Wilson, Marco Gartmann, Jean-Paul Armache, Elizabeth Villa, Leonardo G. Trabuco, Thomas Becker, Thorsten Mielke, Klaus Schulten, Thomas A. Steitz, and Roland Beckmann. Structural insight into nascent polypeptide chain-mediated translational stalling. Science, 326:1412-1415, 2009.
Leonardo G. Trabuco, Elizabeth Villa, Kakoli Mitra, Joachim Frank, and Klaus Schulten. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure, 16:673-683, 2008.
Jen Hsin, James Gumbart, Leonardo G. Trabuco, Elizabeth Villa, Pu Qian, C. Neil Hunter, and Klaus Schulten. Protein-induced membrane curvature investigated through molecular dynamics flexible fitting. Biophys. J., 97:321-329, 2009.
Jens Frauenfeld, James Gumbart, Eli O. van der Sluis, Soledad Funes, Marco Gartmann, Birgitta Beatrix, Thorsten Mielke, Otto Berninghausen, Thomas Becker, Klaus Schulten, and Roland Beckmann. Cryo-EM structure of the ribosome-SecYE complex in the membrane environment. Nat. Struct. Mol. Biol., 18:614-621, 2011.
David E. Tanner, Kwok-Yan Chan, James Phillips, and Klaus Schulten. Parallel generalized Born implicit solvent calculations with NAMD. Journal of Chemical Theory and Computation, 7:3635-3642, 2011.
Kwok-Yan Chan, James Gumbart, Ryan McGreevy, Jean M. Watermeyer, B. Trevor Sewell, and Klaus Schulten. Symmetry-restrained flexible fitting for symmetric EM maps. Structure, 19:1211-1218, 2011.
In-Ja L. Byeon, Xin Meng, Jinwon Jung, Gongpu Zhao, Ruifeng Yang, Jinwoo Ahn, Jiong Shi, Jason Concel, Christopher Aiken, Peijun Zhang, and Angela M. Gronenborn. Structural Convergence between Cryo-EM and NMR Reveals Intersubunit Interactions Critical for HIV-1 Capsid Function. Cell, 139:780-790, 2009.
Gongpu Zhao, Juan R. Perilla, Ernest L. Yufenyuy, Xin Meng, Bo Chen, Jiying Ning, Jinwoo Ahn, Angela M. Gronenborn, Klaus Schulten, Christopher Aiken and Peijun Zhang. Structure of the Mature HIV-1 Capsid by Cryo-EM and All-Atom Molecular Dynamics Simulation. Nature, 497:643-646, 2013.
Laurie Garrett. The Coming Plague. New York, Farrar, Straus and Giroux, 1994.
Wen Li, Gemma C. Atkinson, Nehal S. Thakor, Ülar Allas, Chuao-chao Lu, Kwok-Yan Chan, Tanel Tenson, Klaus Schulten, Kevin S. Wilson, Vasili Hauryliuk, and Joachim Frank. Mechanism of tetracycline resistance by ribosomal protection protein Tet(O). Nature Communications, 4:1477, 2013.
Appendix
Third-party publications employing MDFF
Takeshi Yokoyama, Tanvir R Shaikh, Nobuhiro Iwakura, Hideko Kaji, Akira Kaji and Rajendra K Agrawal. Structural insights into initial and intermediate steps of the ribosome-recycling process. EMBO J., 31:1836-1846, 2012.
Jordi Querol-Audí, Chunli Yan, Xiaojun Xu, Susan E. Tsutakawa, Miaw-Sheue Tsai, John A. Tainer, Priscilla K. Cooper, Eva Nogales and Ivaylo Ivanov. Repair complexes of FEN1 endonuclease, DNA, and Rad9-Hus1-Rad1 are distinguished from their PCNA counterparts by functionally important stability Proc. Natl. Acad. Sci. USA, 109:8528-8533, 2012.
Minghui Li and Wenjun Zheng. All-Atom Structural Investigation of Kinesin-Microtubule Complex Constrained by High-Quality Cryo-Electron-Microscopy Maps Biochemistry, 51:5022-5032, 2012.
Anne-Marie Schönegge, Elizabeth Villa, Friedrich Förster, Reiner Hegerl, Jürgen Peters, Wolfgang Baumeister, Beate Rockel. The Structure of Human Tripeptidyl Peptidase II as Determined by a Hybrid Approach. Structure, 20:593-603, 2012.
Jeff Wereszczynski and J. Andrew McCammon. Simulations of the p97 complex suggest novel conformational states of hydrolysis intermediates. Protein Science, 21:475-486, 2012.
Thomas Becker, Sibylle Franckenberg, Stephan Wickles, Christopher J. Shoemaker, Andreas M. Anger, Jean-Paul Armache, Heidemarie Sieber, Charlotte Ungewickell, Otto Berninghausen, Ingo Daberkow, Annette Karcher, Michael Thomm, Karl-Peter Hopfner, Rachel Green, and Roland Beckmann. Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature, 482:501-506, 2012.
Keren Lasker, Friedrich Förster, Stefan Bohn, Thomas Walzthoeni, Elizabeth Villa, Pia Unverdorbenc, Florian Beck, Ruedi Aebersold, Andrej Sali, and Wolfgang Baumeister. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. PNAS, 109:1380-1387, 2012.
Bethany S. Strunk, Cherisse R. Loucks, Min Su, Harish Vashisth, Shanshan Cheng, Justin Schilling, Charles L. Brooks III, Katrin Karbstein, and Georgios Skinioti. Ribosome Assembly Factors Prevent Premature Translation Initiation by 40S Assembly Intermediates Science, 333:1449-1453, 2011.
Xiao-chen Bai, Xi-jiang Pan, Xiao-jing Wang, Yun-ying Ye, Lei-fu Chang, Dong Leng, Jianlin Lei, Sen-Fang Sui. Characterization of the structure and function of Escherichia coli DegQ as a representative of the DegQ-like proteases of bacterial HtrA family proteins Structure, 19:1328-1337, 2011.
Qiang Guo, Yi Yuan, Yanji Xu, Boya Feng, Liang Liu, Kai Chen, Ming Sun, Zhixiu Yang, Jianlin Lei, and Ning Gao. Structural basis for the function of a small GTPase RsgA on the 30S ribosomal subunit maturation revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA, 108:13100-13105, 2011.
Victor A. Kostyuchenko, Joanita Jakana, Xiangan Liu, Andrew D. Haddow, Myint Aung, Scott C. Weaver, Wah Chiu, and Shee-Mei Lok. The structure of Barmah Forest virus as revealed by cryo-electron microscopy at a 6-Angstrom resolution has detailed transmembrane protein architecture and interactions. J. Virol., 85:9327-9333, 2011.
Petra Wollmann, Sheng Cui, Ramya Viswanathan, Otto Berninghausen, Melissa N. Wells, Manuela Moldt, Gregor Witte, Agata Butryn, Petra Wendler, Roland Beckmann, David T. Auble, and Karl-Peter Hopfner. Structure and mechanism of the Swi2/Snf2 remodeller Mot1 in complex with its substrate TBP. Nature, 475:403-407, 2011.
Aymen S. Yassin, Rajendra K. Agrawal, and Nilesh K. Banavali. Computational exploration of structural hypotheses for an additional sequence in a mammalian mitochondrial protein. PLoS One, 6:e21771, 2011.
Thomas Becker, Jean-Paul Armache, Alexander Jarasch, Andreas M. Anger, Elizabeth Villa, Heidemarie Sieber, Basma Abdel Motaal, Thorsten Mielke, Otto Berninghausen, and Roland Beckmann. Structure of the no-go mRNA decay complex Dom34-Hbs1 bound to a stalled 80S ribosome. Nat. Struct. Mol. Biol., 18:715-720, 2011.
Manjira Ghosh-Kumar, Tanfis I. Alam, Bonnie Draper, John D. Stack, and Venigalla B. Rao. Regulation by interdomain communication of a headful packaging nuclease from bacteriophage T4. Nucleic Acids Res., 39:2742-2755, 2011.
Jean-Paul Armache, Alexander Jarasch, Andreas M. Anger, Elizabeth Villa, Thomas Becker, Shashi Bhushan, Fabrice Jossinet, Michael Habeck, Gülcin Dindar, Sibylle Franckenberg, Viter Marquez, Thorsten Mielke, Michael Thomm, Otto Berninghausen, Birgitta Beatrix, Johannes Söding, Eric Westhof, Daniel N. Wilson, and Roland Beckmann. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-Å resolution. Proc. Natl. Acad. Sci. USA, 107:19748-19753, 2011.
Jean-Paul Armache, Alexander Jarasch, Andreas M. Anger, Elizabeth Villa, Thomas Becker, Shashi Bhushan, Fabrice Jossinet, Michael Habeck, Gülcin Dindar, Sibylle Franckenberg, Viter Marquez, Thorsten Mielke, Michael Thomm, Otto Berninghausen, Birgitta Beatrix, Johannes Södina, Eric Westhof, Daniel N. Wilson, and Roland Beckmann. Localization of eukaryote-specific ribosomal proteins in a 5.5Å cryo-EM map of the 80S eukaryotic ribosome. Proc. Natl. Acad. Sci. USA, 107:19754-19759, 2011.
Félix Weis, Patrick Bron, Emmanuel Giudice, Jean-Paul Rolland, Daniel Thomas, Brice Felden, and Reynald Gillet. tmRNA-SmpB: a journey to the centre of the bacterial ribosome EMBO J., 29:3810-3818, 2010.
Jie Fu, Yaser Hashem, Iwona Wower, Jianlin Lei, Hstau Y Liao, Christian Zwieb, Jacek Wower, and Joachim Frank. Visualizing the transfer-messenger RNA as the ribosome resumes translation. EMBO J., 29:3819-3825, 2010.
Shashi Bhushan, Helge Meyer, Agata L. Starosta, Thomas Becker, Thorsten Mielke, Otto Berninghausen, Michael Sattler, Daniel N. Wilson, and Roland Beckmann. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide Mol. Cell, 40:138-146, 2010.
Michael Lorenz and Kenneth C. Holmes. The actin-myosin interface. Proc. Natl. Acad. Sci. USA, 107:12529-12534, 2010.
Shashi Bhushan, Marco Gartmann, Mario Halic, Jean-Paul Armache, Alexander Jarasch, Thorsten Mielke, Otto Berninghausen, Daniel N Wilson, and Roland Beckmann. α-Helical nascent polypeptide chains visualized within distinct regions of the ribosomal exit tunnel. Nat. Struct. Mol. Biol., 17:313-317, 2010.


